Advanced User Guide - SangerAlignment (AB1)¶
SangerAlignment is in the toppest level of sangeranalyseR (Figure_1), and each SangerAlignment instance corresponds to an alignment of contigs in a Sanger sequencing experiment. Among its slots, there is a SangerContig list which will be aligned into a consensus contig. Users can access to each SangerContig and SangerRead inside a SangerAlignment instance.
In this section, we are going to go through details about a reproducible SangerAlignment analysis example with the AB1 file input in sangeranalyseR. By running the following example codes, you will get an end-to-end SangerAlignment analysis result.
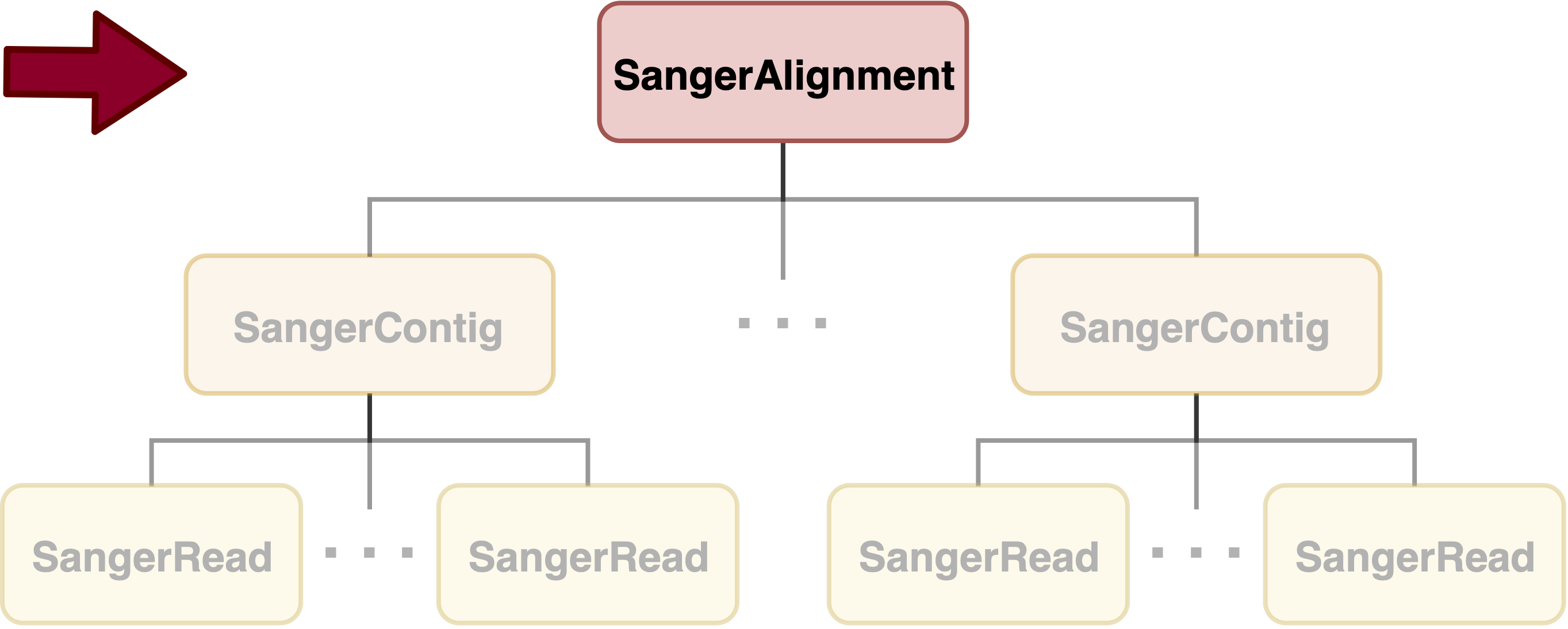
Figure 1. Classes hierarchy in sangeranalyseR, SangerAlignment level.
Preparing SangerAlignment AB1 input¶
The main input file format to create SangerAlignment instance is AB1. Before starting the analysis, users need to prepare one directory containing all AB1 files, and they can be either all placed in the first layer of that directory or be distributed in different subdirectories. In this example, the data are in the sangeranalyseR package; thus, you can simply get its path by running the following codes:
rawDataDir <- system.file("extdata", package = "sangeranalyseR")
parentDir <- file.path(rawDataDir, 'Allolobophora_chlorotica')
The value of parentDir is where all AB1 files are placed. If your operating system is macOS, then its value should look like this:
And we showed the files under parentDir in Figure_2:
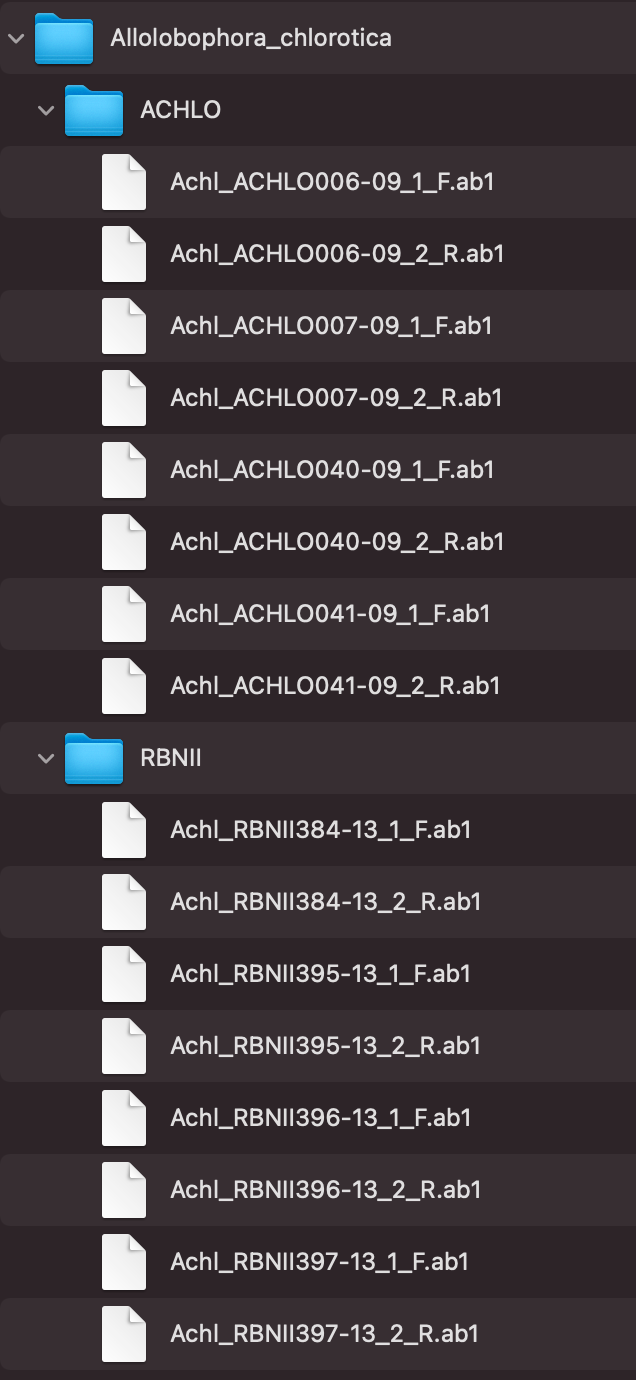
Figure 2. SangerAlignment filename regulation.
Figure_2 shows the file-naming regulation and hierarchy. In this example, Allolobophora_chlorotica is the parent directory, and AB1 files are separated into ACHLO and RBNII directories. There are two ways for users to group their AB1 files which are “regular expression matching” and “CSV file matching”, and following are instructions of how to prepare and name your AB1 input files.
(1) “regular expression matching” SangerAlignment inputs (AB1)¶
For regular expression matching method, sangeranalyseR will group AB1 files based on their contig names and read directions in their filenames automatically; therefore, users have to follow the file-naming regulations below:
Note
- All input files must have .ab1 as its file extension.
- Input files that are in the same contig group must have the same contig name in their filenames.
- Forward or reverse direction has to be specified in the filename.
There are three parameters, ABIF_Directory, REGEX_SuffixForward, and REGEX_SuffixReverse, that define the grouping rule to let sangeranalyseR automatically match correct AB1 files and divide them into forward and reverse directions.
Note
ABIF_Directory: this is the directory that contains all AB1 files, and it can be either an absolute or relative path. We suggest users to put only target AB1 files inside this directory and do not include any other unrelated files.REGEX_SuffixForward: this is a regular expression that matches all filenames in forward direction.greplfunction in R is used.REGEX_SuffixReverse: this is a regular expression that matches all filenames in reverse direction.greplfunction in R is used.
If you don’t know what regular expression is, don’t panic - it’s just a way of recognising text. Please refer to What is a regular expression? for more details. Here is an example of how it works in sangeranalseR:
So how sangeranalyseR works is that it first matches the forward and reverse reads by matching REGEX_SuffixForward and REGEX_SuffixReverse. Then, sangeranalyseR uses the str_split function to split and vectorize their filenames into “contig name” and “direction-suffix” two parts. For those having the same “contig name” will be grouped into the same contig.
Therefore, it is important to have a consistent naming strategy. You need to make sure that AB1 files in the same contig group share the same contig name and carefully select your REGEX_SuffixForward and REGEX_SuffixReverse. The bad file-naming and wrong regex matching might accidentally include reverse reads into the forward read list or vice versa, which will make the program generate wrong results. So, how should we systematically name AB1 files? We suggest users to follow the file-naming regulation in Figure_3.
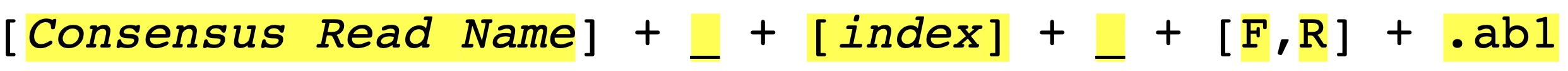
Figure 3. Suggested AB1 file-naming regulation - SangerContig.
As you can see, the first part of the regulation is a consensus read name (or contig name), which helps sangeranalseR to identify which reads should be grouped into the same contig automatically. The second part of the regulation is an index; since there might be more than one read that is in the forward or reverse direction, we recommend you to number your reads in the same contig group. The third part is a direction which is either ‘F’ (forward) or ‘R’ (reverse). Last but not least, files have to end with .ab1 file extension.
To make it more specific, let’s go back to the true example. In Figure_2, there are two subdirectories, ACHLO and RBNII, containing lots of AB1 files from different contigs in the root directory, Allolobophora_chlorotica (ABIF_Directory).
First, we set REGEX_SuffixForward to "_[0-9]*_F.ab1$" and REGEX_SuffixReverse to "_[0-9]*_R.ab1$" to let sangeranalyseR match and group forward and reverse reads automatically. By the regular expression rule, Achl_ACHLO006-09_1_F.ab1, Achl_ACHLO007-09_1_F.ab1, Achl_ACHLO040-09_1_F.ab1, Achl_ACHLO041-09_1_F.ab1, Achl_RBNII384-13_1_F.ab1, Achl_RBNII395-13_1_F.ab1, Achl_RBNII396-13_1_F.ab1, and Achl_RBNII397-13_1_F.ab1 are categorized into forward reads, and Achl_ACHLO006-09_1_R.ab1, Achl_ACHLO007-09_1_R.ab1, Achl_ACHLO040-09_1_R.ab1, Achl_ACHLO041-09_1_R.ab1, Achl_RBNII384-13_1_R.ab1, Achl_RBNII395-13_1_R.ab1, Achl_RBNII396-13_1_R.ab1, and Achl_RBNII397-13_1_R.ab1 are categorized into reverse reads. Then, str_split function is used to split each filename above into “contig name” and “direction-suffix”. Eight contig names are detected in this example which are Achl_ACHLO006-09, Achl_ACHLO007-09, Achl_ACHLO040-09, Achl_ACHLO041-09, Achl_RBNII384-13, Achl_RBNII395-13, Achl_RBNII396-13, and Achl_RBNII397-13. Last, a loop iterates through all contigs, and sangeranalseR creates each of them into a SangerContig instance. You can check Advanced User Guide - SangerContig (AB1) to see how sangeranalyseR creates a SangerContig instance.
The reason why we strongly recommend you to follow this file-naming regulation is that by doing so, you can directly adopt the example regular expression matching values, "_[0-9]*_F.ab1$" and "_[0-9]*_R.ab1$", to group reads and reduce chances of error. Everything mentioned above will be done automatically.
After understanding how parameters work, please refer to Creating SangerAlignment instance from AB1 below to see how sangeranalseR creates SangerAlignment instance.
(2) “CSV file matching” SangerAlignment inputs (AB1)¶
For those who are not familiar with regular expression, we provide a second grouping approach, CSV file matching method. sangeranalyseR will group AB1 files based on the information in a CSV file automatically. The note below shows the regulations:
Note
Here is an example CSV file (Figure 4)

Figure 4. Example CSV file for SangerAlignment instance creation.
- There must be three columns, “reads”, “direction”, and “contig”, in the CSV file.
- The “reads” column stores the filename of AB1 files that are going to be included in the analysis.
- The “direction” column stores the direction of the reads. It must be “F” (forward) or “R” (reverse).
- The “contig” column stores the contig name that each read blongs. Reads in the same contig have to have the same contig name, and they will be grouped into the same contig.
There are two parameters, ABIF_Directory and CSV_NamesConversion,that define the grouping rule to help sangeranalseR to automatically match correct AB1 files and divide them into forward and reverse directions.
Note
ABIF_Directory: this is the directory that contains all AB1 files, and it can be either an absolute or relative path. We suggest users to put only target AB1 files inside this directory and do not include any other unrelated files.CSV_NamesConversion: this is the path to the CSV file. It can be either an absolute or relative path.
The main difference between “CSV file matching” and “regular expression matching” is where the grouping rule is written. For “regular expression matching”, rules are writtein in filenames, and thus more naming requirements are required. In contrast, rules of “CSV file matching” are written in an additional CSV file so it is more flexible on AB1 file-naming.
So how sangeranalyseR works is that it first reads in the CSV file (with “reads”, “direction”, and “contig” columns), find the names of AB1 files listed in “reads”, group them based on “contig”, and assign directions to them based on “direction”.
To make it more specific, let’s go back to the true example. First, we prepare a CSV file (CSV_NamesConversion) and a file directory like Figure_2 (ABIF_Directory) with AB1 files from different contigs. In the CSV file, there are 16 rows and 8 distinct contig names. sangeranalyseR matches “reads” of these 16 rows to filenames in Allolobophora_chlorotica directory. Then sangeranalyseR groups all matched reads, Achl_ACHLO006-09_1_F.ab1, Achl_ACHLO007-09_1_F.ab1, Achl_ACHLO040-09_1_F.ab1, Achl_ACHLO041-09_1_F.ab1, Achl_RBNII384-13_1_F.ab1, Achl_RBNII395-13_1_F.ab1, Achl_RBNII396-13_1_F.ab1, Achl_RBNII397-13_1_F.ab1, Achl_ACHLO006-09_1_R.ab1, Achl_ACHLO007-09_1_R.ab1, Achl_ACHLO040-09_1_R.ab1, Achl_ACHLO041-09_1_R.ab1, Achl_RBNII384-13_1_R.ab1, Achl_RBNII395-13_1_R.ab1, Achl_RBNII396-13_1_R.ab1, and Achl_RBNII397-13_1_R.ab1, into 8 distinct contig names which are Achl_ACHLO006-09, Achl_ACHLO007-09, Achl_ACHLO040-09, Achl_ACHLO041-09, Achl_RBNII384-13, Achl_RBNII395-13, Achl_RBNII396-13, and Achl_RBNII397-13, by the “contig” column. Last, the directions of reads in each contig are assigned by the “direction” column. Take Achl_ACHLO041-09 contig as an example. Its “forward read list” will include Achl_ACHLO041-09_1_F.ab1, and its “reverse read list” will include Achl_ACHLO041-09_1_R.ab1.
After understanding how parameters work, please refer to Creating SangerAlignment instance from AB1 below to see how sangeranalseR creates SangerAlignment instance.
Creating SangerAlignment instance from AB1¶
After preparing the input directory, we can create a SangerAlignment instance by running SangerAlignment constructor function or new method. The constructor function is a wrapper for new method and it makes instance creation more intuitive. Their input parameters are same, and all of them have their default values. For more details about SangerAlignment inputs and slots definition, please refer to sangeranalyseR reference manual. We will explain two SangerAlignment instance creation methods, “regular expression matching” and “CSV file matching”.
(1) “regular expression matching” SangerAlignment creation (AB1)¶
The consturctor function and new method below contain three parameters, ABIF_Directory, REGEX_SuffixForward, and REGEX_SuffixReverse, that we mentioned in the previous section. It also includes important parameters like quality trimming, chromatogram visualization, consensus alignment, contigs alignment, and so on. Run the following code and create my_sangerAlignment instance.
# using `constructor` function to create SangerAlignment instance
my_sangerAlignment <- SangerAlignment(inputSource = "ABIF",
processMethod = "REGEX",
ABIF_Directory = parentDir,
REGEX_SuffixForward = "_[0-9]*_F.ab1$",
REGEX_SuffixReverse = "_[0-9]*_R.ab1$",
TrimmingMethod = "M1",
M1TrimmingCutoff = 0.0001,
M2CutoffQualityScore = NULL,
M2SlidingWindowSize = NULL,
baseNumPerRow = 100,
heightPerRow = 200,
signalRatioCutoff = 0.33,
showTrimmed = TRUE,
refAminoAcidSeq = "SRQWLFSTNHKDIGTLYFIFGAWAGMVGTSLSILIRAELGHPGALIGDDQIYNVIVTAHAFIMIFFMVMPIMIGGFGNWLVPLMLGAPDMAFPRMNNMSFWLLPPALSLLLVSSMVENGAGTGWTVYPPLSAGIAHGGASVDLAIFSLHLAGISSILGAVNFITTVINMRSTGISLDRMPLFVWSVVITALLLLLSLPVLAGAITMLLTDRNLNTSFFDPAGGGDPILYQHLFWFFGHPEVYILILPGFGMISHIISQESGKKETFGSLGMIYAMLAIGLLGFIVWAHHMFTVGMDVDTRAYFTSATMIIAVPTGIKIFSWLATLHGTQLSYSPAILWALGFVFLFTVGGLTGVVLANSSVDIILHDTYYVVAHFHYVLSMGAVFAIMAGFIHWYPLFTGLTLNNKWLKSHFIIMFIGVNLTFFPQHFLGLAGMPRRYSDYPDAYTTWNIVSTIGSTISLLGILFFFFIIWESLVSQRQVIYPIQLNSSIEWYQNTPPAEHSYSELPLLTN",
minReadsNum = 2,
minReadLength = 20,
minFractionCall = 0.5,
maxFractionLost = 0.5,
geneticCode = GENETIC_CODE,
acceptStopCodons = TRUE,
readingFrame = 1,
processorsNum = 2)
# using `new` method to create SangerAlignment instance
my_sangerAlignment <- new("SangerAlignment",
inputSource = "ABIF",
processMethod = "REGEX",
ABIF_Directory = parentDir,
REGEX_SuffixForward = "_[0-9]*_F.ab1$",
REGEX_SuffixReverse = "_[0-9]*_R.ab1$",
TrimmingMethod = "M1",
M1TrimmingCutoff = 0.0001,
M2CutoffQualityScore = NULL,
M2SlidingWindowSize = NULL,
baseNumPerRow = 100,
heightPerRow = 200,
signalRatioCutoff = 0.33,
showTrimmed = TRUE,
refAminoAcidSeq = "SRQWLFSTNHKDIGTLYFIFGAWAGMVGTSLSILIRAELGHPGALIGDDQIYNVIVTAHAFIMIFFMVMPIMIGGFGNWLVPLMLGAPDMAFPRMNNMSFWLLPPALSLLLVSSMVENGAGTGWTVYPPLSAGIAHGGASVDLAIFSLHLAGISSILGAVNFITTVINMRSTGISLDRMPLFVWSVVITALLLLLSLPVLAGAITMLLTDRNLNTSFFDPAGGGDPILYQHLFWFFGHPEVYILILPGFGMISHIISQESGKKETFGSLGMIYAMLAIGLLGFIVWAHHMFTVGMDVDTRAYFTSATMIIAVPTGIKIFSWLATLHGTQLSYSPAILWALGFVFLFTVGGLTGVVLANSSVDIILHDTYYVVAHFHYVLSMGAVFAIMAGFIHWYPLFTGLTLNNKWLKSHFIIMFIGVNLTFFPQHFLGLAGMPRRYSDYPDAYTTWNIVSTIGSTISLLGILFFFFIIWESLVSQRQVIYPIQLNSSIEWYQNTPPAEHSYSELPLLTN",
minReadsNum = 2,
minReadLength = 20,
minFractionCall = 0.5,
maxFractionLost = 0.5,
geneticCode = GENETIC_CODE,
acceptStopCodons = TRUE,
readingFrame = 1,
processorsNum = 2)
In this example, 16 reads are detected and 8 distinct SangerContig instances are created. These SangerContig instances are stored in a “contig list” in my_sangerAlignment, which will be used as the input for the following functions.
Inside the R shell, you can run my_sangerAlignment to get basic information of the instance or run my_sangerAlignment@objectResults@readResultTable to check the creation result of every Sanger read after my_sangerAlignment is successfully created.
Here is the output of my_sangerAlignment:
SangerAlignment S4 instance
Input Source : ABIF
Process Method : REGEX
ABIF Directory : /Library/Frameworks/R.framework/Versions/4.0/Resources/library/sangeranalyseR/extdata/Allolobophora_chlorotica
REGEX Suffix Forward : _[0-9]*_F.ab1$
REGEX Suffix Reverse : _[0-9]*_R.ab1$
Contigs Consensus : TTATAYTTTATTYTRGGCGTCTGAAGCAGGATAGTAGGAGCYGGTATAAGACTCCTAATTCGAATTGAGCTAAGACARCCGGGAGCATTCCTAGGAAGRGATCAACTCTATAACACTATTGTAACTGCTCACGCATTTGTAATAATTTTCTTTCTAGTAATACCTGTATTTATTGGGGGGTTCGGTAATTGACTTCTACCTTTAATACTTGGAGCCCCTGACATGGCATTCCCACGACTTAACAACATAAGATTCTGACTCCTTCCCCCATCACTAATCCTTCTAGTGTCCTCTGCTGCAGTAGAAAAAGGTGCBGGAACTGGATGAACTGTTTATCCRCCCCTAGCAAGAAATATTGCTCATGCCGGCCCATCTGTAGACTTAGCTATYTTTTCTCTTCATTTAGCAGGTGCTTCATCAATCTTAGGKGCYATTAATTTTATYACTACTGTTATTAACATACGATGAAGAGGCTTACGACTTGAACGAATCCCATTATTCGTTTGAGCCGTACTAATTACAGTGGTHCTTCTACTCCTATCYTTACCAGTATTAGCCGGTGCRATTACYATACTACTTACCGATCGAAATCTAAATACCTCCTTCTTTGAYCCTGCTGGAGGTGGAGATCCCATCCTCTACCAACACTTATTCTGATTTTTTGGTCACCCTGAG
SUCCESS [2021-13-07 23:16:16] 'SangerAlignment' is successfully created!
Here is the output of my_sangerAlignment@objectResults@readResultTable:
readName creationResult errorType errorMessage inputSource direction
1 Achl_ACHLO006-09_1_F.ab1 TRUE None None ABIF Forward Read
2 Achl_ACHLO006-09_2_R.ab1 TRUE None None ABIF Reverse Read
3 Achl_ACHLO007-09_1_F.ab1 TRUE None None ABIF Forward Read
4 Achl_ACHLO007-09_2_R.ab1 TRUE None None ABIF Reverse Read
5 Achl_ACHLO040-09_1_F.ab1 TRUE None None ABIF Forward Read
6 Achl_ACHLO040-09_2_R.ab1 TRUE None None ABIF Reverse Read
7 Achl_ACHLO041-09_1_F.ab1 TRUE None None ABIF Forward Read
8 Achl_ACHLO041-09_2_R.ab1 TRUE None None ABIF Reverse Read
9 Achl_RBNII384-13_1_F.ab1 TRUE None None ABIF Forward Read
10 Achl_RBNII384-13_2_R.ab1 TRUE None None ABIF Reverse Read
11 Achl_RBNII395-13_1_F.ab1 TRUE None None ABIF Forward Read
12 Achl_RBNII395-13_2_R.ab1 TRUE None None ABIF Reverse Read
13 Achl_RBNII396-13_1_F.ab1 TRUE None None ABIF Forward Read
14 Achl_RBNII396-13_2_R.ab1 TRUE None None ABIF Reverse Read
15 Achl_RBNII397-13_1_F.ab1 TRUE None None ABIF Forward Read
16 Achl_RBNII397-13_2_R.ab1 TRUE None None ABIF Reverse Read
(2) “CSV file matching” SangerAlignment creation (AB1)¶
The consturctor function and new method below contain two parameters, ABIF_Directory, and CSV_NamesConversion, that we mentioned in the previous section. It also includes important parameters like quality trimming, chromatogram visualization, consensus alignment, contigs alignment, and so on. Run the following code and create my_sangerAlignment instance.
csv_namesConversion <- file.path(rawDataDir, "ab1", "SangerAlignment", "names_conversion_all.csv")
# using `constructor` function to create SangerAlignment instance
my_sangerAlignment <- SangerAlignment(inputSource = "ABIF",
processMethod = "CSV",
ABIF_Directory = parentDir,
CSV_NamesConversion = csv_namesConversion,
TrimmingMethod = "M1",
M1TrimmingCutoff = 0.0001,
M2CutoffQualityScore = NULL,
M2SlidingWindowSize = NULL,
baseNumPerRow = 100,
heightPerRow = 200,
signalRatioCutoff = 0.33,
showTrimmed = TRUE,
refAminoAcidSeq = "SRQWLFSTNHKDIGTLYFIFGAWAGMVGTSLSILIRAELGHPGALIGDDQIYNVIVTAHAFIMIFFMVMPIMIGGFGNWLVPLMLGAPDMAFPRMNNMSFWLLPPALSLLLVSSMVENGAGTGWTVYPPLSAGIAHGGASVDLAIFSLHLAGISSILGAVNFITTVINMRSTGISLDRMPLFVWSVVITALLLLLSLPVLAGAITMLLTDRNLNTSFFDPAGGGDPILYQHLFWFFGHPEVYILILPGFGMISHIISQESGKKETFGSLGMIYAMLAIGLLGFIVWAHHMFTVGMDVDTRAYFTSATMIIAVPTGIKIFSWLATLHGTQLSYSPAILWALGFVFLFTVGGLTGVVLANSSVDIILHDTYYVVAHFHYVLSMGAVFAIMAGFIHWYPLFTGLTLNNKWLKSHFIIMFIGVNLTFFPQHFLGLAGMPRRYSDYPDAYTTWNIVSTIGSTISLLGILFFFFIIWESLVSQRQVIYPIQLNSSIEWYQNTPPAEHSYSELPLLTN",
minReadsNum = 2,
minReadLength = 20,
minFractionCall = 0.5,
maxFractionLost = 0.5,
geneticCode = GENETIC_CODE,
acceptStopCodons = TRUE,
readingFrame = 1,
processorsNum = 1)
# using `new` method to create SangerAlignment instance
my_sangerAlignment <- new("SangerAlignment",
processMethod = "CSV",
ABIF_Directory = parentDir,
CSV_NamesConversion = csv_namesConversion,
TrimmingMethod = "M1",
M1TrimmingCutoff = 0.0001,
M2CutoffQualityScore = NULL,
M2SlidingWindowSize = NULL,
baseNumPerRow = 100,
heightPerRow = 200,
signalRatioCutoff = 0.33,
showTrimmed = TRUE,
refAminoAcidSeq = "SRQWLFSTNHKDIGTLYFIFGAWAGMVGTSLSILIRAELGHPGALIGDDQIYNVIVTAHAFIMIFFMVMPIMIGGFGNWLVPLMLGAPDMAFPRMNNMSFWLLPPALSLLLVSSMVENGAGTGWTVYPPLSAGIAHGGASVDLAIFSLHLAGISSILGAVNFITTVINMRSTGISLDRMPLFVWSVVITALLLLLSLPVLAGAITMLLTDRNLNTSFFDPAGGGDPILYQHLFWFFGHPEVYILILPGFGMISHIISQESGKKETFGSLGMIYAMLAIGLLGFIVWAHHMFTVGMDVDTRAYFTSATMIIAVPTGIKIFSWLATLHGTQLSYSPAILWALGFVFLFTVGGLTGVVLANSSVDIILHDTYYVVAHFHYVLSMGAVFAIMAGFIHWYPLFTGLTLNNKWLKSHFIIMFIGVNLTFFPQHFLGLAGMPRRYSDYPDAYTTWNIVSTIGSTISLLGILFFFFIIWESLVSQRQVIYPIQLNSSIEWYQNTPPAEHSYSELPLLTN",
minReadsNum = 2,
minReadLength = 20,
minFractionCall = 0.5,
maxFractionLost = 0.5,
geneticCode = GENETIC_CODE,
acceptStopCodons = TRUE,
readingFrame = 1,
processorsNum = 1)
First, you need to load the CSV file into the R environment. If you are still don’t know how to prepare it, please check (2) “CSV file matching” SangerAlignment inputs (AB1). Then, it will follow rules in the CSV file and create my_sangerAlignment. After it’s created, inside the R shell, you can run my_sangerAlignment to get basic information of the instance or run my_sangerAlignment@objectResults@readResultTable to check the creation result of every Sanger read after my_sangerAlignment is successfully created.
Here is the output of my_sangerAlignment:
SangerAlignment S4 instance
Input Source : ABIF
Process Method : CSV
ABIF Directory : /Library/Frameworks/R.framework/Versions/4.0/Resources/library/sangeranalyseR/extdata/Allolobophora_chlorotica
CSV Names Conversion : /Library/Frameworks/R.framework/Versions/4.0/Resources/library/sangeranalyseR/extdata/ab1/SangerAlignment/names_conversion_all.csv
Contigs Consensus : TTATAYTTTATTYTRGGCGTCTGAAGCAGGATAGTAGGAGCYGGTATAAGACTCCTAATTCGAATTGAGCTAAGACARCCGGGAGCATTCCTAGGAAGRGATCAACTCTATAACACTATTGTAACTGCTCACGCATTTGTAATAATTTTCTTTCTAGTAATACCTGTATTTATTGGGGGGTTCGGTAATTGACTTCTACCTTTAATACTTGGAGCCCCTGACATGGCATTCCCACGACTTAACAACATAAGATTCTGACTCCTTCCCCCATCACTAATCCTTCTAGTGTCCTCTGCTGCAGTAGAAAAAGGTGCBGGAACTGGATGAACTGTTTATCCRCCCCTAGCAAGAAATATTGCTCATGCCGGCCCATCTGTAGACTTAGCTATYTTTTCTCTTCATTTAGCAGGTGCTTCATCAATCTTAGGKGCYATTAATTTTATYACTACTGTTATTAACATACGATGAAGAGGCTTACGACTTGAACGAATCCCATTATTCGTTTGAGCCGTACTAATTACAGTGGTHCTTCTACTCCTATCYTTACCAGTATTAGCCGGTGCRATTACYATACTACTTACCGATCGAAATCTAAATACCTCCTTCTTTGAYCCTGCTGGAGGTGGAGATCCCATCCTCTACCAACACTTATTCTGATTTTTTGGTCACCCTGAG
SUCCESS [2021-14-07 01:48:28] 'SangerAlignment' is successfully created!
Here is the output of my_sangerAlignment@objectResults@readResultTable:
readName creationResult errorType errorMessage inputSource direction
1 Achl_ACHLO006-09_1_F.ab1 TRUE None None ABIF Forward Read
2 Achl_ACHLO006-09_2_R.ab1 TRUE None None ABIF Reverse Read
3 Achl_ACHLO007-09_1_F.ab1 TRUE None None ABIF Forward Read
4 Achl_ACHLO007-09_2_R.ab1 TRUE None None ABIF Reverse Read
5 Achl_ACHLO040-09_1_F.ab1 TRUE None None ABIF Forward Read
6 Achl_ACHLO040-09_2_R.ab1 TRUE None None ABIF Reverse Read
7 Achl_ACHLO041-09_1_F.ab1 TRUE None None ABIF Forward Read
8 Achl_ACHLO041-09_2_R.ab1 TRUE None None ABIF Reverse Read
9 Achl_RBNII384-13_1_F.ab1 TRUE None None ABIF Forward Read
10 Achl_RBNII384-13_2_R.ab1 TRUE None None ABIF Reverse Read
11 Achl_RBNII395-13_1_F.ab1 TRUE None None ABIF Forward Read
12 Achl_RBNII395-13_2_R.ab1 TRUE None None ABIF Reverse Read
13 Achl_RBNII396-13_1_F.ab1 TRUE None None ABIF Forward Read
14 Achl_RBNII396-13_2_R.ab1 TRUE None None ABIF Reverse Read
15 Achl_RBNII397-13_1_F.ab1 TRUE None None ABIF Forward Read
16 Achl_RBNII397-13_2_R.ab1 TRUE None None ABIF Reverse Read
Updating SangerAlignment quality trimming parameters¶
In the previous Creating SangerAlignment instance from AB1 part, the constructor function will apply the quality trimming parameters to all reads. After creating a SangerAlignment S4 instance, users can change the trimming parameters by running updateQualityParam function which will update all reads with the new trimming parameters and redo reads alignment in SangerContig and contigs alignment in SangerAlignment. If users want to do quality trimming read by read instead all at once, please read Launching SangerAlignment Shiny app.
newSangerAlignment <- updateQualityParam(my_sangerAlignment,
TrimmingMethod = "M2",
M1TrimmingCutoff = NULL,
M2CutoffQualityScore = 29,
M2SlidingWindowSize = 15)
Launching SangerAlignment Shiny app¶
We create an interactive local Shiny app for users to go into each SangerRead and SangerContig in SangerAlignment instance. Users only need to run one function with previously created instance as input, my_sangerAlignment, and the SangerAlignment Shiny app will pop up. Here, we will go through pages in the three levels.
launchApp(my_sangerAlignment)
SangerAlignment page (SA app)¶
Figure 5 is the initial page and the toppest layer of SangerAlignment App. It provides basic parameters in SangerAlignment instance, contigs alignment result and phylogenetic tree etc. Before checking the results, users need to click “Re-calculate Contigs Alignment” button to do contigs alignment in order to get the updated results. From the left-hand side panel, we can clearly see the hierarchy of the SangerAlignment S4 instance and easily access to all reads and contigs in it.
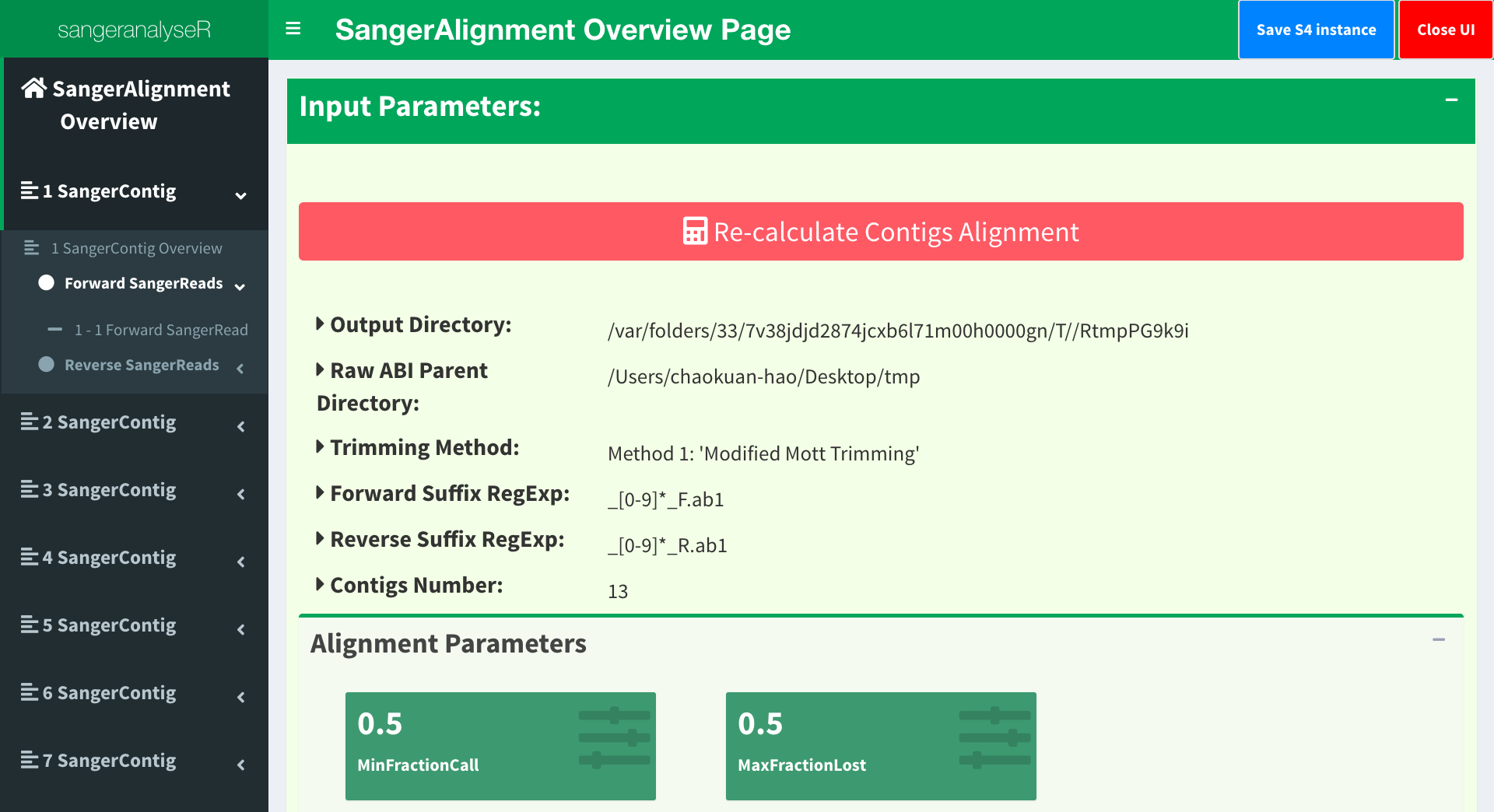
Figure 5. SangerAlignment Shiny app initial page - SangerAlignment Page.
Scroll down a bit, users can see the contigs alignment result generated by DECIPHER R package embedded in SangerAlignment page. Figure 6 shows the contigs alignment result.
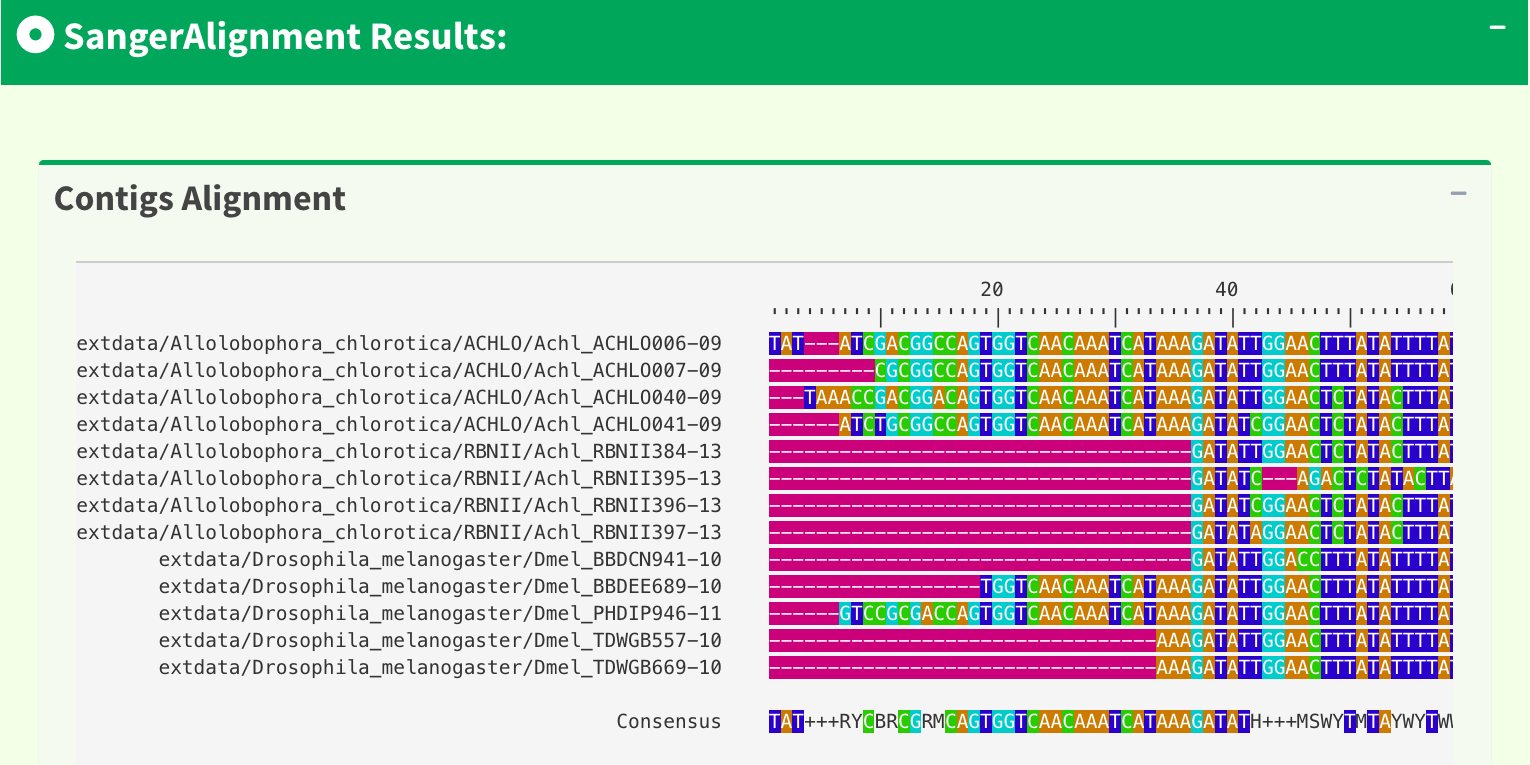
Figure 6. SangerAlignment Page - contigs alignment result.
In SangerAlignment page, the phylogenetic tree result is provided as well (Figure 7). The tree is generated by ape R package which uses neighbor-joining algorithm.

Figure 7. SangerAlignment Page - phylogenetic tree result.
SangerContig page (SA app)¶
Now, let’s go to the page in the next level, SangerContig page. Users can click into all contigs and check their results. Figure 8 shows the overview page of Contig 1. Notice that there is a red “Re-calculate Contig” button. After changing the quality trimming parameters, users need to click the button before checking the results below in order to get the updated information.
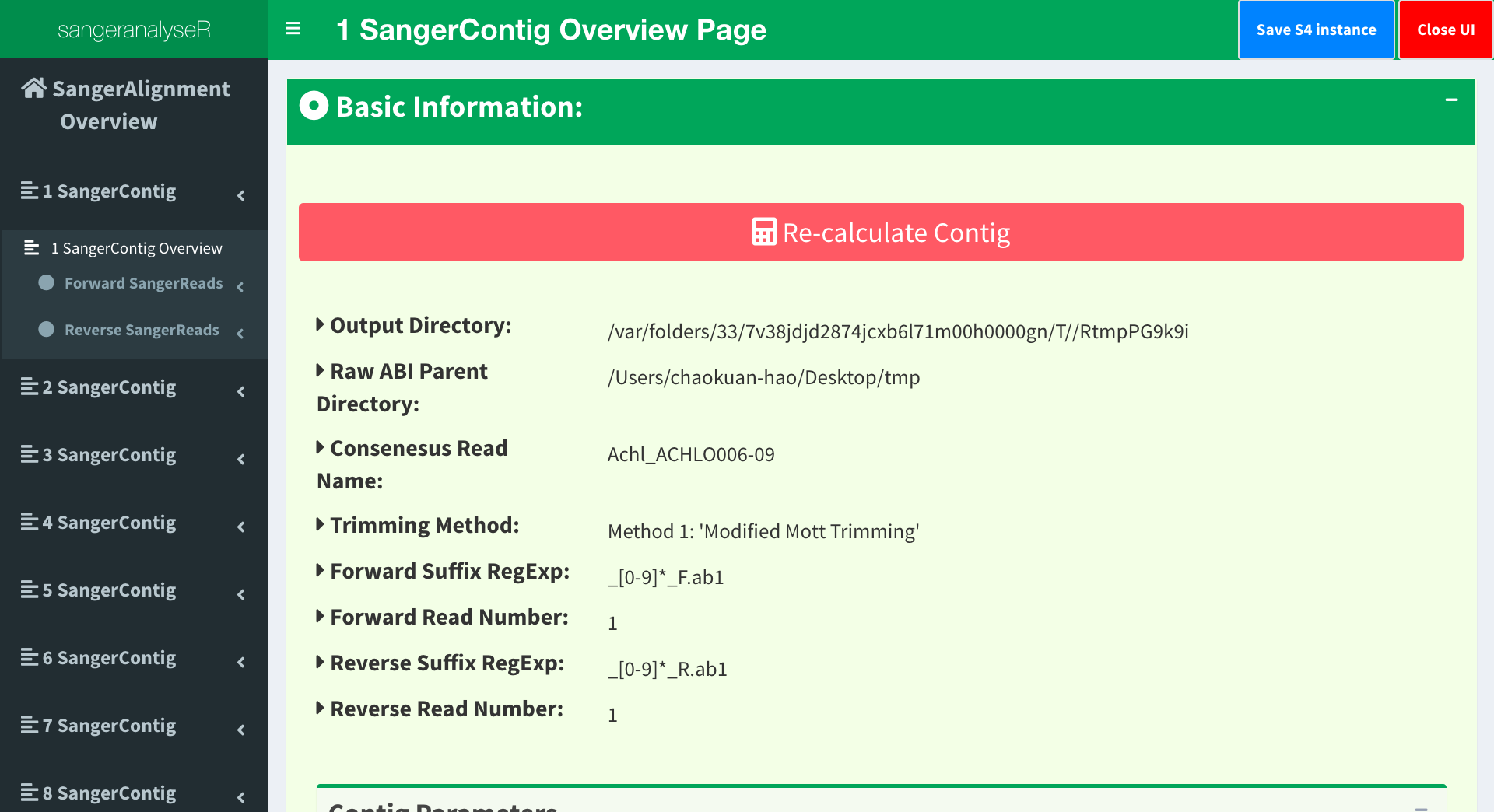
Figure 8. SangerAlignment Shiny app - SangerContig page.
The information provided in this page includes : “input parameters”, “genetic code table”, “reference amino acid sequence”, “reads alignment”, “difference data frame”, “dendrogram”, “sample distance heatmap”, “indels data frame”, “stop codons data frame”. Figure 9 and Figure 10 show part of the results in the SangerContig page. The results are dynamic based on the trimming parameters from user inputs.
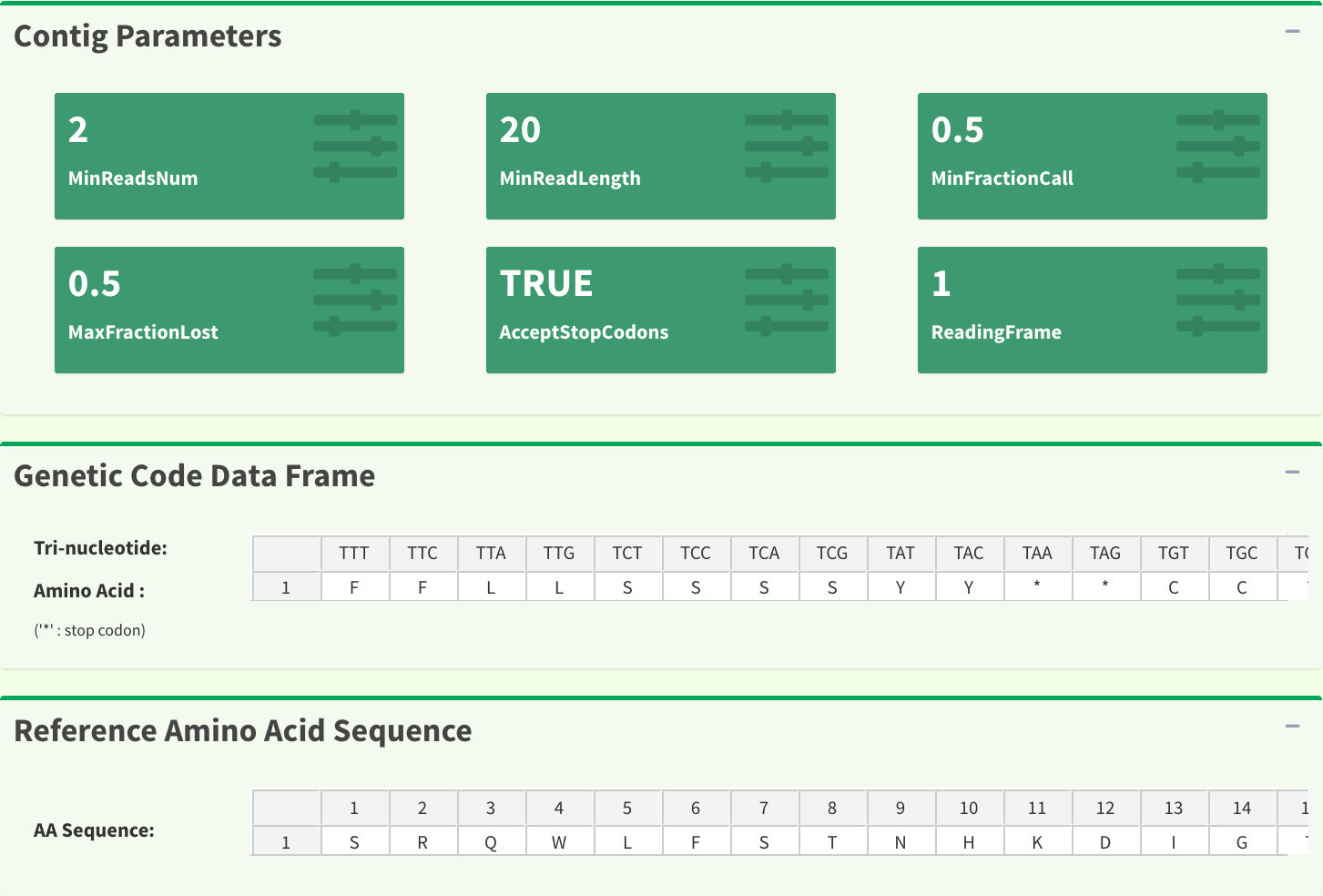
Figure 9. SangerContig page - contig-related parameters, genetic code and reference amino acid sequence.
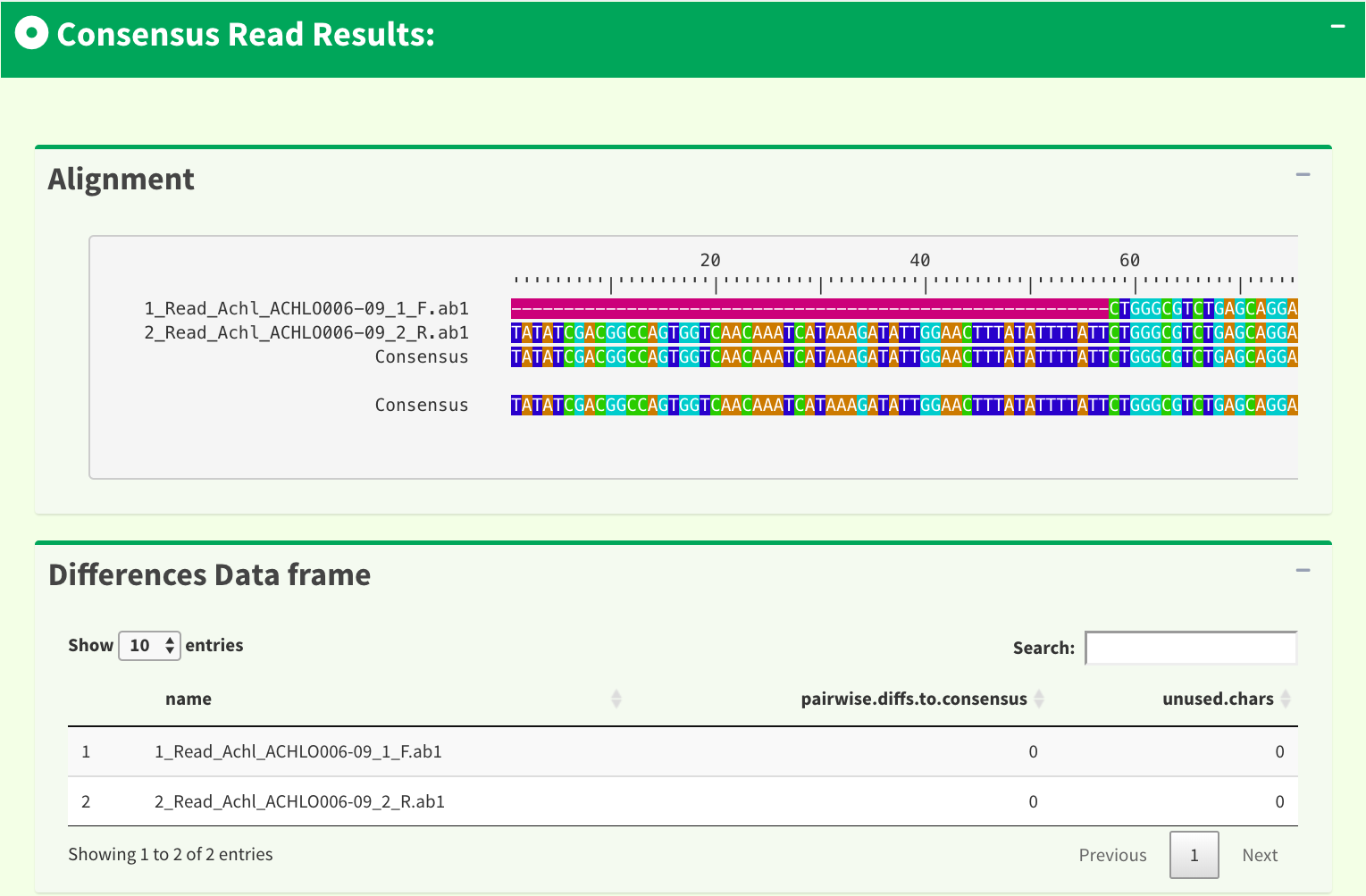
Figure 10. SangerContig page - reads alignment and difference data frame.
SangerRead page (SA app)¶
Now, let’s go to the page in the lowest level, SangerRead page. SangerRead page contains all details of a read including its trimming and chromatogram inputs and results. All reads are in “forward” or “reverse” direction. Under “Contig Overview” tab (SangerContig page), there are two expendable tabs, “Forward Reads” and “Reverse Reads” storing corresponding reads on the left-hand side navigation panel in Figure 11. In this example, there are one read in each tab and Figure 11 shows the “1 - 1 Forward Read” page. It provides basic information, quality trimming inputs, chromatogram plotting inputs etc. Primary/secondary sequences in this figure are dynamic based on the signalRatioCutoff value for base calling and the length of them are always same. Another thing to mention is that primary/secondary sequences and the sequences in the chromatogram in Figure 16 below will always be same after trimming and their color codings for A/T/C/G are same as well.
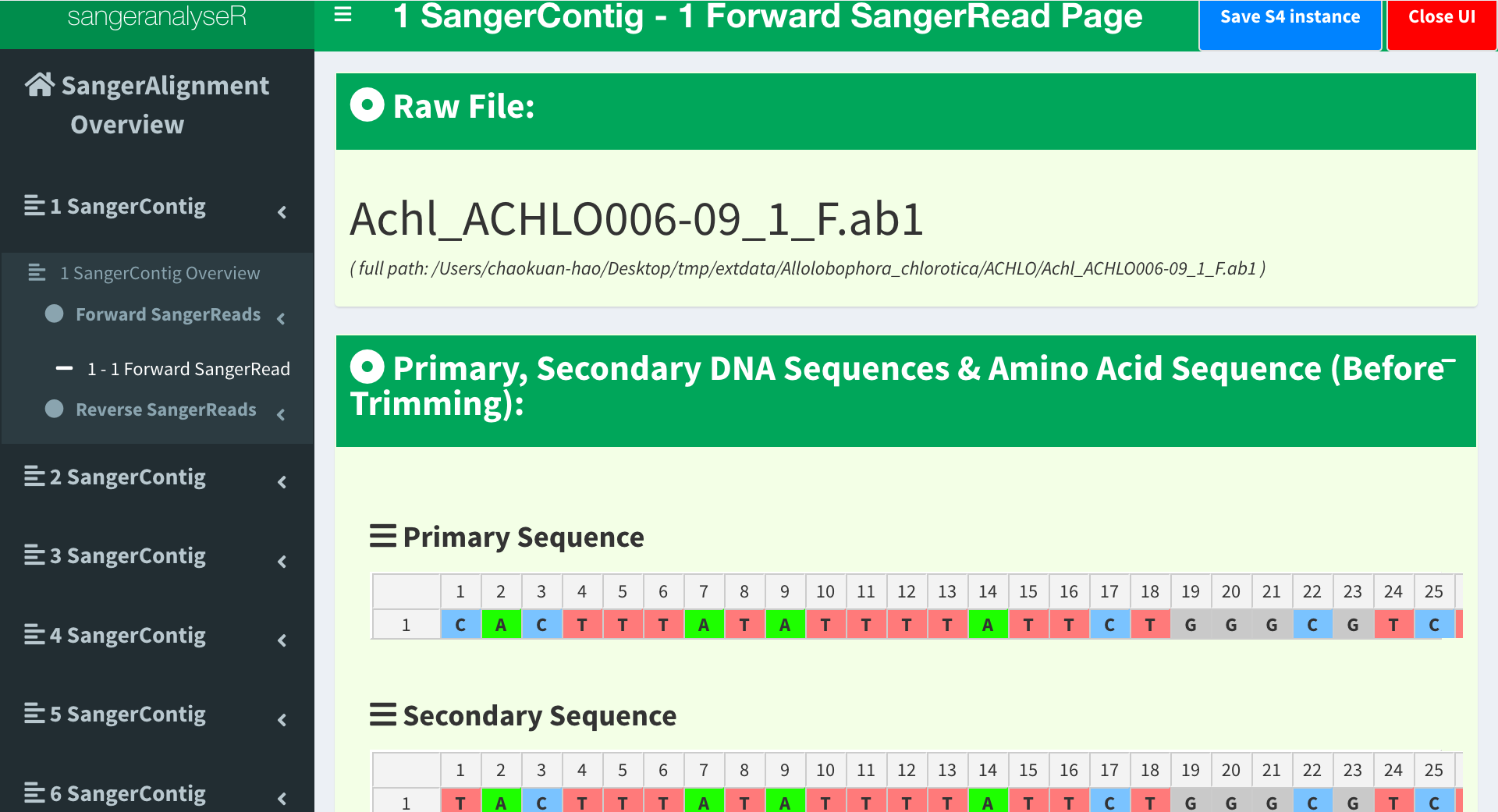
Figure 11. SangerAlignment Shiny app - SangerRead page.
In quality trimming steps, we removes fragment at both ends of sequencing reads with low quality score. It is important because trimmed reads will improves alignment results. Figure 12 shows the UI for Trimming Method 1 (M1): ‘Modified Mott Trimming’. This method is implemented in Phred. Users can change the cutoff score and click “Apply Trimming Parameters” button to update the UI. The value of input must be between 0 and 1. If the input is invalid, the cutoff score will be set to default 0.0001.
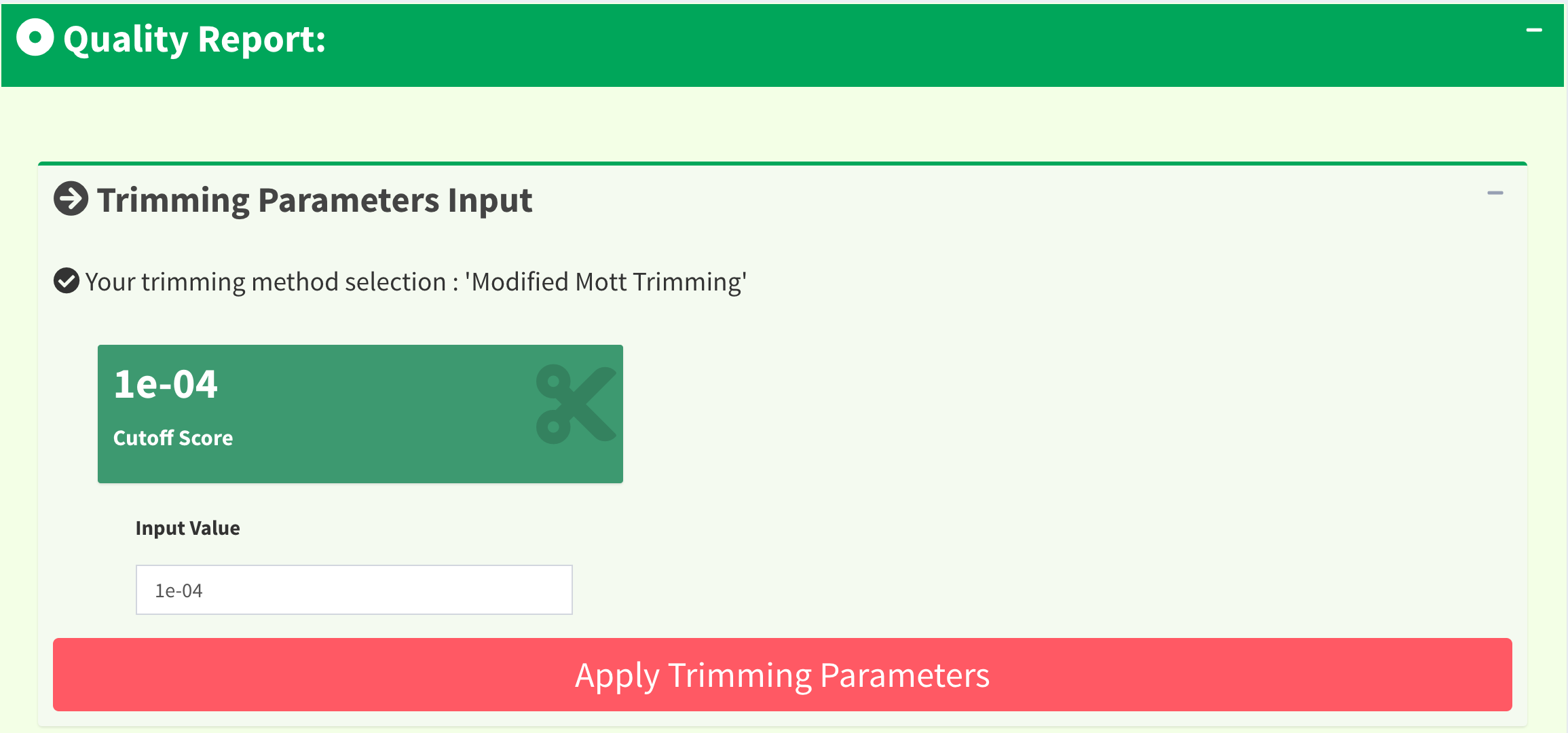
Figure 12. SangerRead page - Trimming Method 1 (M1): ‘Modified Mott Trimming’ UI.
Figure 13 shows another quality trimming methods for users to choose from, Trimming Method 2 (M2): ‘Trimmomatics Sliding Window Trimming’. This method is implemented in Trimmomatics. Users can change the cutoff quality score as well as sliding window size and click “Apply Trimming Parameters” button to update the UI. The value of cutoff quality score must be between 0 and 60 (default 20); the value of sliding window size must be between 0 and 40 (default 10). If the inputs are invalid, their values will be set to default.
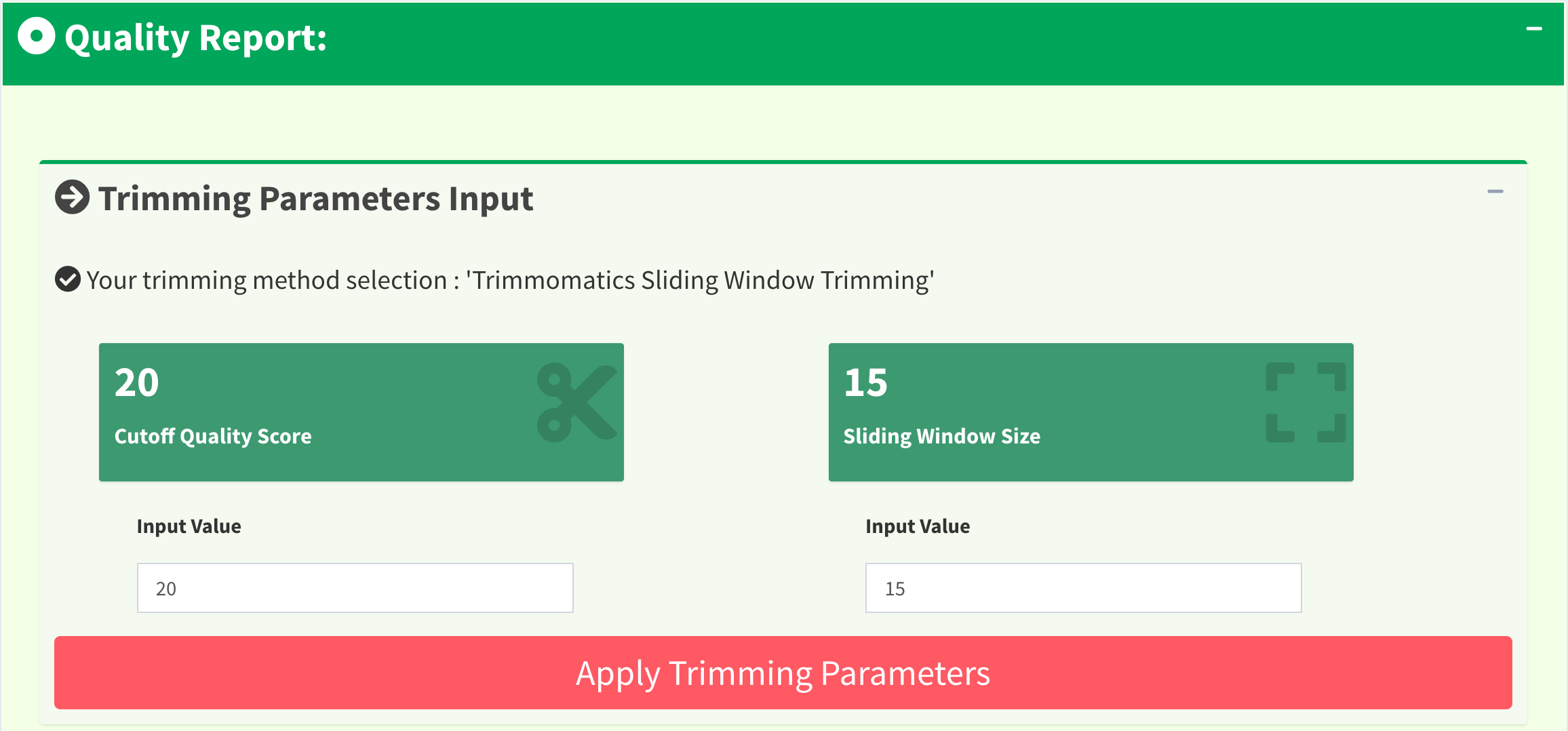
Figure 13. SangerRead page - Trimming Method 2 (M2): ‘Trimmomatics Sliding Window Trimming’ UI.
Figure 14 shows the quality report before and after trimming. After clicking the “Apply Trimming Parameters” button, the values of these information boxes will be updated to the latest values.

Figure 14. SangerRead page - read quality report before / after trimming.
In Figure 15, the x-axis is the index of the base pairs; the y-axis is the Phred quality score. The green horizontal bar at the top of the plot is the raw read region and the orange horizontal bar represents the trimmed read region. Both Figure 15 trimming plot and Figure 16 chromatogram will be updated once users change the quality trimming parameters and click the “Apply Trimming Parameters” button in Figure 16.
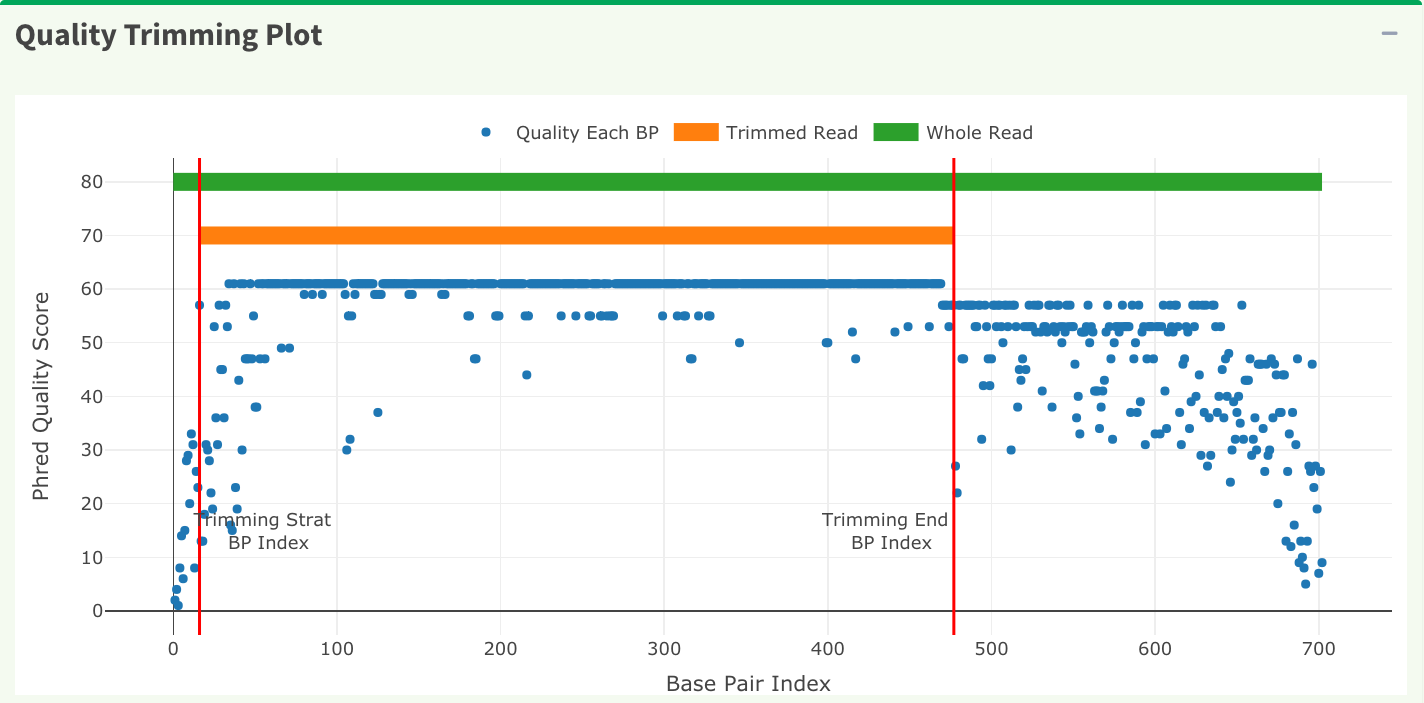
Figure 15. SangerRead page - quality trimming plot.
If we only see primary and secondary sequences in the table, we will loose some variations. Chromatogram is very helpful to check the peak resolution. Figure 16 shows the panel of plotting chromatogram. Users can change four parameters: Base Number Per Row, Height Per Row, Signal Ratio Cutoff, and Show Trimmed Region. Among them, Signal Ratio Cutoff is the key parameter. If its value is default value 0.33, it indicates that the lower peak should be at least 1/3rd as high as the higher peak for it count as a secondary peak.
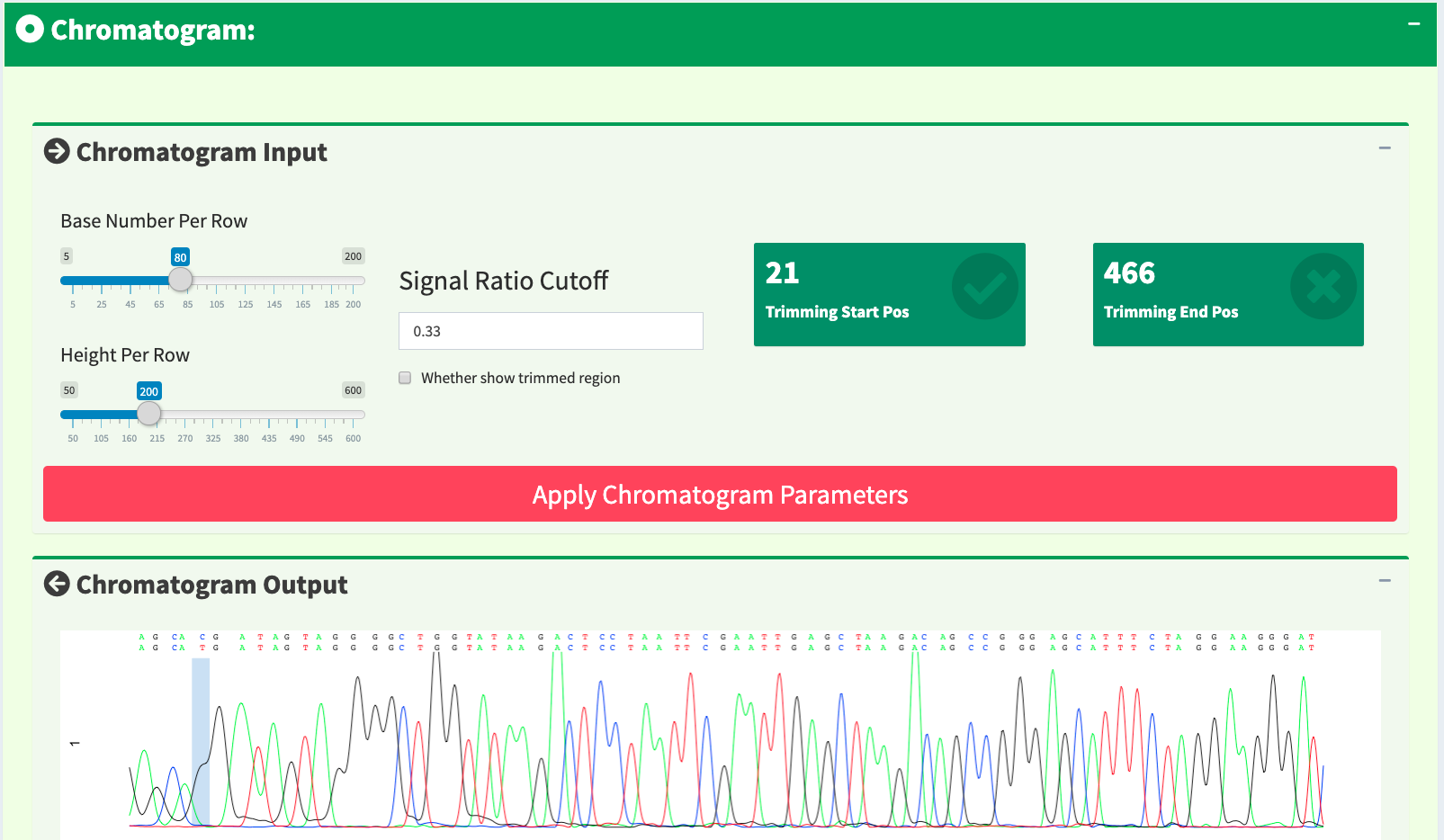
Figure 16. SangerRead page - chromatogram panel.
Here is an example of applying new chromatogram parameters. We click “Show Trimmed Region” to set its value from FALSE to TRUE. Figure 17 shows the loading notification popup during base calling and chromatogram plotting.
Figure 17. SangerRead page - loading notification popup during replotting chromatogram.
After replotting the chromatogram, trimmed region is showed in red striped region. Figure 18 shows part of the the chromatogram (1 bp ~ 240 bp). Moreover, chromatogram will be replotted when trimmed positions or chromatogram parameters are updated.
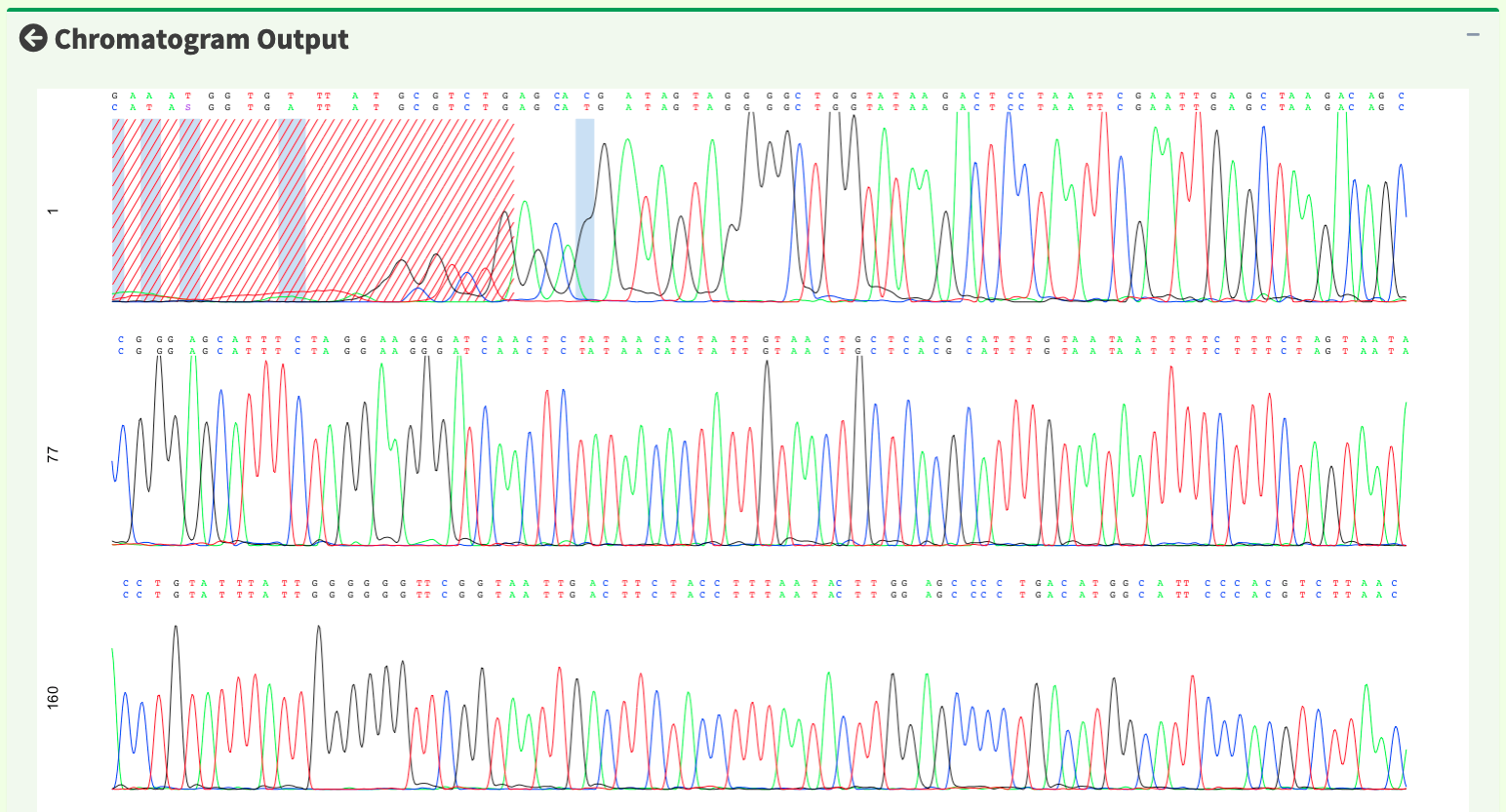
Figure 18. SangerRead page - chromatogram with trimmed region showed.
To let users browse the trimmed primary/secondary sequences without finding “Trimming Start Point” and “Trimming End Point” by themselves, we provide the final trimmed primary/secondary sequences that will be used for reads alignment in table format with quality scores in Figure 19. Frameshift amino acid sequences are also provided.
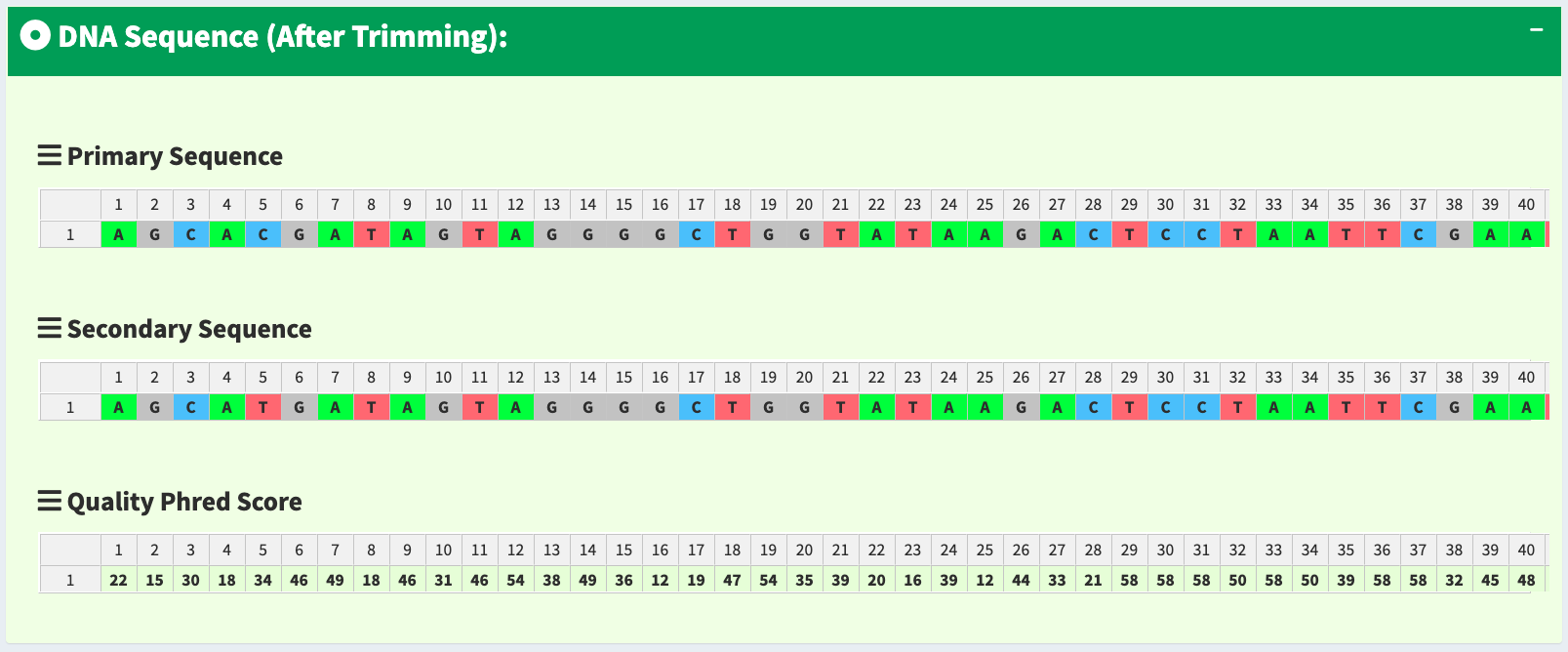
Figure 19. SangerRead page - trimmed primary/secondary sequences and Phred quality score in table format.
We have updated the trimming and chromatogram parameters for each read. Now, we need to click “Re-calculate contig” button to do alignment again. Last but not least, we can save all data into a new ‘SangerContig’ S4 instance by clicking “Save S4 instance button”. New S4 instance will be saved in Rda format. Users can run readRDS function to load it into current R environment. Figure 20 shows some hints in the save notification popup.

Figure 20. SangerRead page - saving notification popup.
Writing SangerAlignment FASTA files (AB1)¶
Users can write the SangerAlignment instance, my_sangerAlignment, to FASTA files. There are four options for users to choose from in selection parameter.
contigs_unalignment: Writing contigs into a single FASTA file.contigs_alignment: Writing contigs alignment and contigs consensus read to a single FASTA file.all_reads: Writing all reads to a single FASTA file.all: Writing contigs, contigs alignment, and all reads into three different files.
Below is the oneliner for writing out FASTA files. This function mainly depends on writeXStringSet function in Biostrings R package. Users can set the compression level through writeFasta function.
writeFasta(my_sangerAlignment,
outputDir = tempdir(),
compress = FALSE,
compression_level = NA,
selection = "all")
Users can download the output FASTA file of this example through the following three links:
Generating SangerAlignment report (AB1)¶
Last but not least, users can save SangerAlignment instance, my_sangerAlignment, into a report after the analysis. The report will be generated in HTML by knitting Rmd files.
Users can set includeSangerContig and includeSangerRead parameters to decide to which level the SangerAlignment report will go. Moreover, after the reports are generated, users can easily navigate through reports in different levels within the HTML file.
One thing to pay attention to is that if users have many reads, it will take quite a long time to write out all reports. If users only want to generate the contig result, remember to set includeSangerRead and includeSangerContig to FALSE in order to save time.
generateReport(my_sangerAlignment,
outputDir = tempdir(),
includeSangerRead = FALSE,
includeSangerContig = FALSE)
Here is the generated SangerAlignment html report of this example (ABIF). Users can access to ‘Basic Information’, ‘Contigs Consensus’, ‘Contigs Alignment’, ‘Contigs Tree’, and ‘Contig Reports’ sections inside it. Furthermore, users can also navigate through html reports of all contigs and forward and reverse SangerRead in this SangerAlignment report.
Code summary (SangerAlignment, AB1)¶
(1) Preparing SangerAlignment AB1 inputs¶
rawDataDir <- system.file("extdata", package = "sangeranalyseR")
parentDir <- file.path(rawDataDir, 'Allolobophora_chlorotica')
(2) Creating SangerAlignment instance from AB1¶
(2.1) “Regular Expression Method” SangerAlignment creation (AB1)¶
# using `constructor` function to create SangerAlignment instance
my_sangerAlignment <- SangerAlignment(inputSource = "ABIF",
processMethod = "REGEX",
ABIF_Directory = parentDir,
REGEX_SuffixForward = "_[0-9]*_F.ab1$",
REGEX_SuffixReverse = "_[0-9]*_R.ab1$",
refAminoAcidSeq = "SRQWLFSTNHKDIGTLYFIFGAWAGMVGTSLSILIRAELGHPGALIGDDQIYNVIVTAHAFIMIFFMVMPIMIGGFGNWLVPLMLGAPDMAFPRMNNMSFWLLPPALSLLLVSSMVENGAGTGWTVYPPLSAGIAHGGASVDLAIFSLHLAGISSILGAVNFITTVINMRSTGISLDRMPLFVWSVVITALLLLLSLPVLAGAITMLLTDRNLNTSFFDPAGGGDPILYQHLFWFFGHPEVYILILPGFGMISHIISQESGKKETFGSLGMIYAMLAIGLLGFIVWAHHMFTVGMDVDTRAYFTSATMIIAVPTGIKIFSWLATLHGTQLSYSPAILWALGFVFLFTVGGLTGVVLANSSVDIILHDTYYVVAHFHYVLSMGAVFAIMAGFIHWYPLFTGLTLNNKWLKSHFIIMFIGVNLTFFPQHFLGLAGMPRRYSDYPDAYTTWNIVSTIGSTISLLGILFFFFIIWESLVSQRQVIYPIQLNSSIEWYQNTPPAEHSYSELPLLTN")
# using `new` method to create SangerAlignment instance
my_sangerAlignment <- new("SangerAlignment",
inputSource = "ABIF",
processMethod = "REGEX",
ABIF_Directory = parentDir,
REGEX_SuffixForward = "_[0-9]*_F.ab1$",
REGEX_SuffixReverse = "_[0-9]*_R.ab1$",
refAminoAcidSeq = "SRQWLFSTNHKDIGTLYFIFGAWAGMVGTSLSILIRAELGHPGALIGDDQIYNVIVTAHAFIMIFFMVMPIMIGGFGNWLVPLMLGAPDMAFPRMNNMSFWLLPPALSLLLVSSMVENGAGTGWTVYPPLSAGIAHGGASVDLAIFSLHLAGISSILGAVNFITTVINMRSTGISLDRMPLFVWSVVITALLLLLSLPVLAGAITMLLTDRNLNTSFFDPAGGGDPILYQHLFWFFGHPEVYILILPGFGMISHIISQESGKKETFGSLGMIYAMLAIGLLGFIVWAHHMFTVGMDVDTRAYFTSATMIIAVPTGIKIFSWLATLHGTQLSYSPAILWALGFVFLFTVGGLTGVVLANSSVDIILHDTYYVVAHFHYVLSMGAVFAIMAGFIHWYPLFTGLTLNNKWLKSHFIIMFIGVNLTFFPQHFLGLAGMPRRYSDYPDAYTTWNIVSTIGSTISLLGILFFFFIIWESLVSQRQVIYPIQLNSSIEWYQNTPPAEHSYSELPLLTN")
(2.2) “CSV file matching” SangerAlignment creation (AB1)¶
csv_namesConversion <- file.path(rawDataDir, "ab1", "SangerAlignment", "names_conversion_all.csv")
# using `constructor` function to create SangerAlignment instance
my_sangerAlignment <- SangerAlignment(inputSource = "ABIF",
processMethod = "CSV",
ABIF_Directory = parentDir,
CSV_NamesConversion = csv_namesConversion,
refAminoAcidSeq = "SRQWLFSTNHKDIGTLYFIFGAWAGMVGTSLSILIRAELGHPGALIGDDQIYNVIVTAHAFIMIFFMVMPIMIGGFGNWLVPLMLGAPDMAFPRMNNMSFWLLPPALSLLLVSSMVENGAGTGWTVYPPLSAGIAHGGASVDLAIFSLHLAGISSILGAVNFITTVINMRSTGISLDRMPLFVWSVVITALLLLLSLPVLAGAITMLLTDRNLNTSFFDPAGGGDPILYQHLFWFFGHPEVYILILPGFGMISHIISQESGKKETFGSLGMIYAMLAIGLLGFIVWAHHMFTVGMDVDTRAYFTSATMIIAVPTGIKIFSWLATLHGTQLSYSPAILWALGFVFLFTVGGLTGVVLANSSVDIILHDTYYVVAHFHYVLSMGAVFAIMAGFIHWYPLFTGLTLNNKWLKSHFIIMFIGVNLTFFPQHFLGLAGMPRRYSDYPDAYTTWNIVSTIGSTISLLGILFFFFIIWESLVSQRQVIYPIQLNSSIEWYQNTPPAEHSYSELPLLTN")
# using `new` method to create SangerAlignment instance
my_sangerAlignment <- new("SangerAlignment",
processMethod = "CSV",
ABIF_Directory = parentDir,
CSV_NamesConversion = csv_namesConversion,
refAminoAcidSeq = "SRQWLFSTNHKDIGTLYFIFGAWAGMVGTSLSILIRAELGHPGALIGDDQIYNVIVTAHAFIMIFFMVMPIMIGGFGNWLVPLMLGAPDMAFPRMNNMSFWLLPPALSLLLVSSMVENGAGTGWTVYPPLSAGIAHGGASVDLAIFSLHLAGISSILGAVNFITTVINMRSTGISLDRMPLFVWSVVITALLLLLSLPVLAGAITMLLTDRNLNTSFFDPAGGGDPILYQHLFWFFGHPEVYILILPGFGMISHIISQESGKKETFGSLGMIYAMLAIGLLGFIVWAHHMFTVGMDVDTRAYFTSATMIIAVPTGIKIFSWLATLHGTQLSYSPAILWALGFVFLFTVGGLTGVVLANSSVDIILHDTYYVVAHFHYVLSMGAVFAIMAGFIHWYPLFTGLTLNNKWLKSHFIIMFIGVNLTFFPQHFLGLAGMPRRYSDYPDAYTTWNIVSTIGSTISLLGILFFFFIIWESLVSQRQVIYPIQLNSSIEWYQNTPPAEHSYSELPLLTN")
(3) Updating SangerAlignment quality trimming parameters (AB1)¶
newSangerAlignment <- updateQualityParam(my_sangerAlignment,
TrimmingMethod = "M2",
M1TrimmingCutoff = NULL,
M2CutoffQualityScore = 29,
M2SlidingWindowSize = 15)
(5) Writing SangerAlignment FASTA files (AB1)¶
writeFasta(my_sangerAlignment)
You will get three FASTA files:
(6) Generating SangerAlignment report (AB1)¶
generateReport(my_sangerAlignment)
You can check the html report of this SangerAlignment example (ABIF).