Advanced User Guide - SangerRead (AB1)¶
SangerRead is in the bottommost level of sangeranalyseR (Figure_1), and each SangerRead object corresponds to a single read (one AB1 file) in a Sanger sequencing experiment. SangerRead class extends sangerseq class from sangerseqR package and contains input parameters and results of quality trimming and chromatogram. In this section, we are going to go through detailed sangeranalyseR data analysis steps in SangerRead level with AB1 file input.
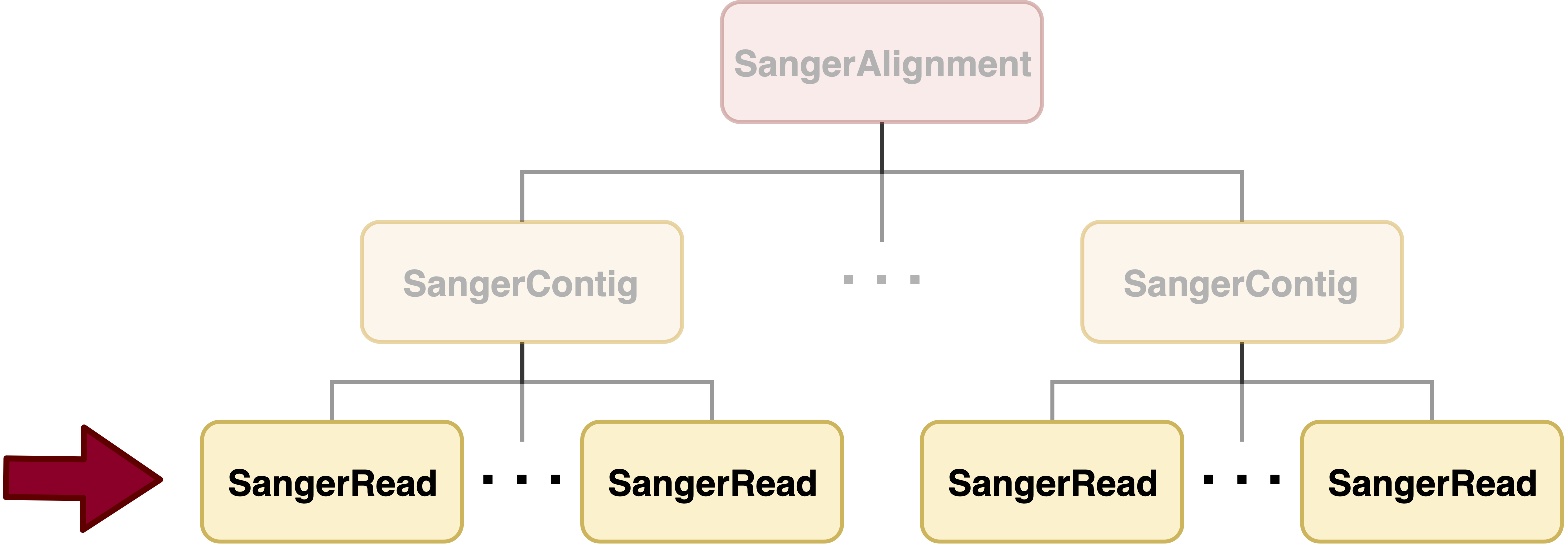
Figure 1. Hierarchy of classes in sangeranalyseR, SangerRead level.
Preparing SangerRead AB1 input¶
The main input file format to create SangerRead instance is AB1. Before starting the analysis, users need to prepare one target AB1 file, and in this example, it is in the sangeranalyseR package; thus, you can simply get its path by running the following codes:
inputFilesPath <- system.file("extdata/", package = "sangeranalyseR")
A_chloroticaFFN <- file.path(inputFilesPath,
"Allolobophora_chlorotica",
"ACHLO",
"Achl_ACHLO006-09_1_F.ab1")
The only hard regulation of the filename, Achl_ACHLO006-09_1_F.ab1 in this example, is that the input file must have .ab1 as its file extension. There are some suggestions about the filename in the note below:
Note
- AB1 file should be indexed for better consistency with file-naming regulation for SangerContig and SangerAlignment.
- Forward or reverse direction should be specified in the filename.
Figure_2 shows the suggested file-naming strategy. The filename should contain four main parts: “Contig name”, “Index number”, “Direction” and “ab1 file extension”.
- “Contig name” :
Achl_RBNII397-13 - “Index number” :
1 - “Direction” :
F - “ab1 file extension” :
.ab1
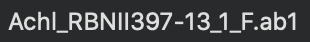
Figure 2. SangerRead filename regulation.
In SangerRead section, it is not compulsory to follow the file-naming regulation because users can directly specify the filename in input (see Creating SangerRead instance from AB1); however, in the SangerContig and SangerAlignment, sangeranalyseR will automatically group files, so it is compulsory to have systematic file-naming strategy. For more details, please read Advanced User Guide - SangerContig (AB1) and Advanced User Guide - SangerAlignment (AB1). Figure_3 shows the suggested AB1 file-naming regulation.
Figure 3. Suggested AB1 file-naming regulation - SangerRead.
Creating SangerRead instance from AB1¶
After preparing the SangerRead input AB1 file, A_chloroticaFFN , the next step is to create a SangerRead instance by running SangerRead constructor function or new method. The constructor function is a wrapper for the new method which makes instance creation more intuitive. The inputs include Basic Parameters, Trimming Parameters, and Chromatogram Parameters, and all of them have default values. In the example below, we show both SangerRead creation methods with important parameters.
# using `constructor` function to create SangerRead instance
sangerReadF <- SangerRead(readFeature = "Forward Read",
readFileName = A_chloroticaFFN,
geneticCode = GENETIC_CODE,
TrimmingMethod = "M1",
M1TrimmingCutoff = 0.0001,
M2CutoffQualityScore = NULL,
M2SlidingWindowSize = NULL,
baseNumPerRow = 100,
heightPerRow = 200,
signalRatioCutoff = 0.33,
showTrimmed = TRUE)
# using `new` method to create SangerRead instance
sangerReadF <- new("SangerRead",
readFeature = "Forward Read",
readFileName = A_chloroticaFFN,
geneticCode = GENETIC_CODE,
TrimmingMethod = "M1",
M1TrimmingCutoff = 0.0001,
M2CutoffQualityScore = NULL,
M2SlidingWindowSize = NULL,
baseNumPerRow = 100,
heightPerRow = 200,
signalRatioCutoff = 0.33,
showTrimmed = TRUE)
The inputs of SangerRead constructor function and new method are the same. For more details about SangerRead inputs and slots definition, please refer to the sangeranalyseR reference manual. The created SangerRead instance, sangerReadF, is used as the input for the following functions.
Inside the R shell, you can run sangerReadF to get basic information of the instance or run sangerReadF@objectResults@readResultTable to check the creation result of every Sanger read after sangerReadF is successfully created.
Here is the output of sangerReadF:
SangerRead S4 instance
Input Source : ABIF
Read Feature : Forward Read
Read FileName : Achl_ACHLO006-09_1_F.ab1
Trimming Method : M1
Primary Sequence : CTGGGCGTCTGAGCAGGAATGGTTGGAGCCGGTATAAGACTTCTAATTCGAATCGAGCTAAGACAACCAGGAGCGTTCCTGGGCAGAGACCAACTATACAATACTATCGTTACTGCACACGCATTTGTAATAATCTTCTTTCTAGTAATGCCTGTATTCATCGGGGGATTCGGAAACTGGCTTTTACCTTTAATACTTGGAGCCCCCGATATAGCATTCCCTCGACTCAACAACATGAGATTCTGACTACTTCCCCCATCACTGATCCTTTTAGTGTCCTCTGCGGCGGTAGAAAAAGGCGCTGGTACGGGGTGAACTGTTTATCCGCCTCTAGCAAGAAATCTTGCCCACGCAGGCCCGTCTGTAGATTTAGCCATCTTTTCCCTTCATTTAGCGGGTGCGTCTTCTATTCTAGGGGCTATTAATTTTATCACCACAGTTATTAATATGCGTTGAAGAGG
Secondary Sequence : CTGGGCGTCTGAGCAGGAATGGTTGGAGCCGGTATAAGACTTCTAATTCGAATCGAGCTAAGACAACCAGGAGCGTTCCTGGGCAGAGACCAACTATACAATACTATCGTTACTGCACACGCATTTGTAATAATCTTCTTTCTAGTAATGCCTGTATTCATCGGGGGATTCGGAAACTGGCTTTTACCTTTAATACTTGGAGCCCCCGATATAGCATTCCCTCGACTCAACAACATGAGATTCTGACTACTTCCCCCATCACTGATCCTTTTAGTGTCCTCTGCGGCGGTAGAAAAAGGCGCTGGTACGGGGTGAACTGTTTATCCGCCTCTAGCAAGAAATCTTGCCCACGCAGGCCCGTCTGTAGATTTAGCCATCTTTTCCCTTCATTTAGCGGGTGCGTCTTCTATTCTAGGGGCTATTAATTTTATCACCACAGTTATTAATATGCGTTGAAGAGG
SUCCESS [2021-12-07 23:31:16] 'Achl_ACHLO006-09_1_F.ab1' is successfully created!
Here is the output of sangerReadF@objectResults@readResultTable:
readName creationResult errorType errorMessage inputSource direction
1 Achl_ACHLO006-09_1_F.ab1 TRUE None None ABIF Forward Read
Visualizing SangerRead trimmed read¶
Before going to Writing SangerRead FASTA file (AB1) and Generating SangerRead report (AB1) pages, it is suggested to visualize the trimmed SangerRead. Run the qualityBasePlot function to get the result in Figure_4. It shows the quality score for each base pairs and the trimming start/end points of the sequence.
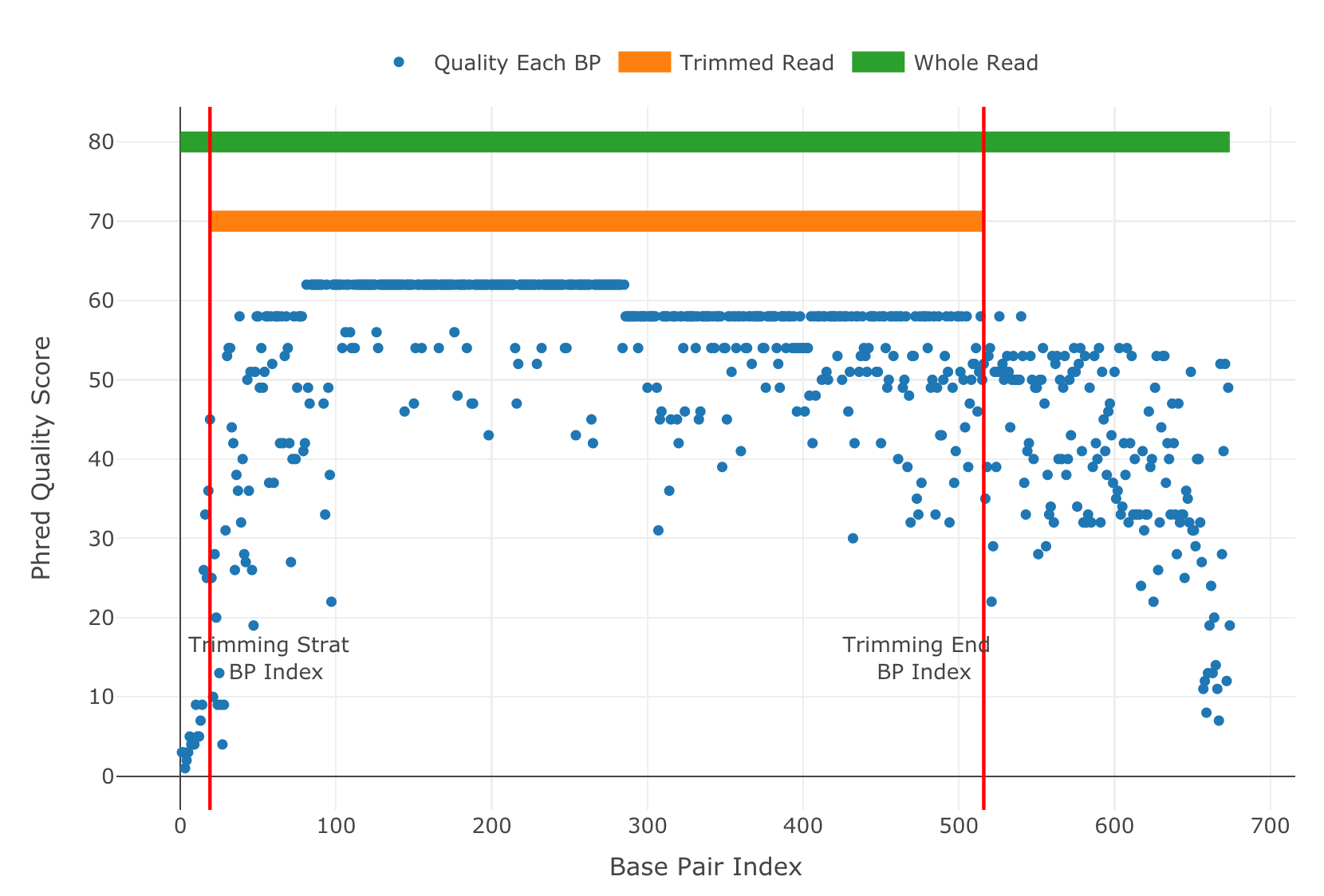
Figure 4. SangerRead trimmed read visualization.
qualityBasePlot(sangerReadF)
Updating SangerRead quality trimming parameters¶
In the previous Creating SangerRead instance from AB1 part, the constructor function applies the quality trimming parameters to the read. These parameters are not fixed. After instance creation, users can run updateQualityParam function which will change the QualityReport instance inside the SangerRead and update frameshift amino acid sequences.
newSangerRead <- updateQualityParam(sangerReadF,
TrimmingMethod = "M2",
M1TrimmingCutoff = NULL,
M2CutoffQualityScore = 29,
M2SlidingWindowSize = 15)
Writing SangerRead FASTA file (AB1)¶
After quality trimming, users can write sangerReadF into a FASTA file. Below is the one-liner that needs to be run. This function, writeFasta, mainly depends on writeXStringSet function in Biostrings R package. Users can further set the compression level through it.
writeFasta(sangerReadF,
outputDir = tempdir(),
compress = FALSE,
compression_level = NA)
Users can download the output FASTA file of this example.
Generating SangerRead report (AB1)¶
Last but not least, users can save sangerReadF into a static HTML report by knitting Rmd files. In this example, tempdir function will generate a random path.
generateReport(sangerReadF,
outputDir = tempdir())
SangerRead_Report_ab1.html is the generated SangerRead report html of this example. Users can access to ‘Basic Information’, ‘DNA Sequence’, ‘Amino Acids Sequence’, ‘Quality Trimming’ and ‘Chromatogram’ sections inside this report.
Code summary (SangerRead, ab1)¶
(1) Preparing SangerRead AB1 input¶
inputFilesPath <- system.file("extdata/", package = "sangeranalyseR")
A_chloroticaFFN <- file.path(inputFilesPath,
"Allolobophora_chlorotica",
"ACHLO",
"Achl_ACHLO006-09_1_F.ab1")
(2) Creating SangerRead instance from AB1¶
# using `constructor` function to create SangerRead instance
sangerReadF <- SangerRead(readFeature = "Forward Read",
readFileName = A_chloroticaFFN)
# using `new` method to create SangerRead instance
sangerReadF <- new("SangerRead",
readFeature = "Forward Read",
readFileName = A_chloroticaFFN)
(4) Writing SangerRead FASTA file (AB1)¶
writeFasta(sangerReadF)
And you will get one FASTA file:
(5) Generating SangerRead report (AB1)¶
generateReport(sangerReadF)
You can check the html report of this SangerRead example (ABIF).